Процесс отщепления карбоксильной группы аминокислот в виде СO2 получил название декарбоксилирования. Несмотря на ограниченный круг субстратов (аминокислот и их производных), подвергающихся декарбоксилированию в животных тканях, образующиеся продукты реакции (названные биогенными аминами) обладают сильным фармакологическим действием на множество физиологических функций человека и животных. В животных тканях показано декарбоксилирование следующих аминокислот и их производных: тирозина, триптофана, 5-окситриптофана, валина, серина, гистидина, глутаминовой и γ-оксиглутаминовой кислот, 3, 4-диоксифенилаланина, цистеина и цистеин-сульфиновой кислоты, аргинина, орнитина, S-аденозилметионина и α-аминомалоновой кислоты. Помимо этого, у микроорганизмов и растений открыто декарбоксилирование этих и ряда других аминокислот. Сведения о декарбоксилировании аминокислот в живых организмах суммированы в табл. 39 [показать] .
| Таблица 39. Ферментативное декарбоксилированне аминокислот и их производных | ||||
| Субстрат | Продукт реакции | Распространение | ||
| животные | растения | микроорганизмы | ||
| S-Аденозил-метионин | S-Аденозилгомоцистеамин | + | + | |
| Парааминобенэойная кислота | Анилин | + | ||
| α-Аминомалоновая кислота | Глицин | + | ||
| α-Аминомасляная кислота | Пропиламин | + | ||
| Антраниловая кислота | Анилин | + | ||
| L-Аргинин | Агматин | + | ||
| L-Аспарагиновая кислота | β-Аланин | + (?) | + | |
| L-Аспарагиновая кислота | α-Аланин | + | ||
| L-Валин | 2-Метилпропиламин | + | + | + |
| L-Гистидин | Гистамин | + | + | |
| Две молекулы глицина | 2CO2+2NH3+CH3COOH | + | ||
| L-Глутаминовая кислота | γ-Аминомасляная кислота | + | + | + |
| Мезо-α; ε-диаминопимелиновая кислота | L-Лизин | + | ||
| 3,4- Диоксифенилаланин | 3,4-Диоксифенилэтиламин | + | + | + |
| 3,4-Диоксифенилсерин | Норадреналин | + | ||
| L-Изолейцин | 2-Метилбутиламин | + | + | |
| L-Лейцин | 3-Метилбутиламин | + | + | |
| L-Лизин | Кадаверин | + | ||
| γ-Метилен-L-глутаминовая кислота | γ-Амино-α-метиленмасляная кислота | + | ||
| Норвалин | н-Бутиламин | + | ||
| Алло-β-оксиглутаминовая кислота | γ-Амино-α-оксимасляная кислота | + | ||
| γ-Оксиглутаминовая кислота | α-oкси-γ-аминомасляная кислота | + | + | |
| 5-Оксилиэин | 2-оксикадаверин | + | ||
| 5-Окситриптофан | Серотонин | + | ||
| n-Оксифенилсерин | n-Оксифениламиноэтанол | + | ||
| L-Орнитин | Путресцин | + | + | |
| L-Серин | Этаноламин | + | ||
| L-Тирозин | Тирамин | + | + | + |
| L-Триптофан | Триптамин | + | ||
| L-Фенилаланин | Фенилэтиламин | + | + | |
| L-Цистеиновая кислота | Таурин | + | ||
| L-Цистеинсульфиновая кислота | Гипотаурин | + | ||
Общая схема процесса декарбоксилирования аминокислот может быть представлена в следующем виде:
В живых организмах открыто четыре типа декарбоксилирования аминокислот.
- α-Декарбоксилирование, характерное для большинства природных аминокислот и их производных, при котором отщепляется карбоксильная группа, стоящая по соседству с α-углеродным атомом. Продуктами реакции являются СО2 и биогенные амины:
R-CH(NH2)-COOH —> R-CH2-NH2 + CO2
ω-Декарбоксилированне, характерное для микроорганизмов. Например, из аспарагиновой кислоты этим путем образуется α-аланин:
НООС-СН2-CH(NH2)-СООН —> СН3-CH(NH2)-СООН + СО2
Декарбоксилирование, связанное с реакцией трансаминирования:

В этой реакции образуются альдегид и новая аминокислота, соответствующая исходной кетокислоте.
Декарбоксилирование, связанное с реакцией конденсации двух молекул:

Эта реакция в тканях животных осуществляется при синтезе δ-аминолевулиновой кислоты из глицина и сукцинил-КоА (см. Синтез гемоглобина) и при синтезе 3-кетосфинганина (сфинголипидов), а также у растений при синтезе биотина.
| Помимо этих реакций, у Peptococcus glycinophilus открыта еще одна реакция декарбоксилирования, сопряженная с генерацией энергии (синтезом АТФ). Этот анаэробный организм утилизирует глицин в качестве единственного источника углерода, превращая его в ацетат: В этом уравнении реакции участвует множество ферментов, а также тетрагидрофолиевая кислота (ТГФ); первая часть уравнения включает декарбоксилирование одной молекулы глицина: Эта последняя реакция также катализируется комплексом ферментов, одним из которых является пиридоксальфосфатзависимая глициндекарбоксилаза. Аналогичный путь катаболизма глицина доказан (без генерации энергии) в митохондриях печени крыс; таким образом, открыт еще один путь образования одноуглеродных фрагментов, используемых организмом для множества синтетических реакций. |
Реакции декарбоксилирования в отличие от других процессов промежуточного обмена аминокислот являются необратимыми. Они катализируются специфическими ферментами — декарбоксилазами аминокислот, отличающимися от декарбоксилаз α-кетокислот как по белковому компоненту, так и по природе кофермента. Декарбоксилазы аминокислот состоят из белковой части, обеспечивающей специфичность действия, и простетической группы, представленной пиридоксальфосфатом, как и у трансаминаз.
Таким образом, в двух совершенно различных процессах аминокислот участвует один и тот же кофермент- пиридоксальфосфат. Исключение составляют две декарбоксилазы — гистидиндекарбоксилаза Micrococcus и Lactobacillus и S-аденозилметиониндекарбоксилаза Е. coli, содержащие вместо пиридоксальфосфата остаток пировиноградной кислоты (С. Р. Мардашев, Снелл). Соответствующие декарбоксилазы животных тканей содержат пиридоксальфосфат.

Механизм реакции декарбоксилирования аминокислот в соответствии с общей теорией пиридоксалевого катализа сводится к образованию пиридоксальфосфат-субстратного комплекса в активном центре фермента (представленного, как и в реакциях трансаминирования, шиффовым основанием пиридоксальфосфата и аминокислоты, см. формулу).
Образование подобного комплекса в сочетании с некоторым оттягиванием электронов белковой частью молекулы декарбоксилазы сопровождается лабилизацией связей «а», «б» и «в», благодаря которой аминокислота приобретает способность к различного рода превращениям (декарбоксилирование, трансаминирование, дегидратация и т. д.).
Ниже будут представлены отдельные примеры декарбоксилирования аминокислот (и соответствующих декарбоксилаз), в частности тех аминокислот, продукты реакции которых обладают мощным фармакологическим действием. Одним из хорошо изученных ферментов является декарбоксилаза ароматических аминокислот, не обладающая строгой субстратной специфичностью и катализирующая декарбоксилирование L-изомеров триптофана, 5-гидрокситриптофана и 3,4-диоксифенилаланина (ДОФА); продуктами реакций, помимо СO2, являются соответственно триптамин, серотонин и диоксифенилэтиламин (дофамин):

Декарбоксилаза ароматических аминокислот получена в чистом виде (мол. м. 112 000); кофермент-пиридоксальфосфат. В больших количествах она содержится в надпочечниках и ЦНС. Она играет важную роль в регуляции синтеза биогенных аминов. Образующийся из триптофана под действием этого фермента продукт — триптамин — наделен сосудосуживающим действием.
Другим, более изученным, биогенным амином, образующимся из 5-гидрокситриптофана, является 5-гидрокситриптамин, или серотонин. Помимо сосудосуживающего действия, серотонин участвует в центральной регуляции артериального давления, температуры тела, дыхания и почечной фильтрации. Он является медиатором нервных процессов в ЦНС. Некоторые авторы считают серотонин причастным к развитию аллергии, демпинг-синдрома, токсикоза беременности, карциноидного синдрома и геморрагических диатезов.

Относительно третьего продукта декарбоксилазной реакции — дофамина — следует прежде всего указать на ферментные системы и промежуточные продукты, ведущие к его образованию. Это важно, так как дофамин является предшественником катехоламинов (норадреналина и адреналина). Источником ДОФА в организме является тирозин, который под действием специфической гидроксилазы превращается в 3,4-диоксифенилаланин. Тирозингидроксилаза открыта в надпочечниках, в ткани мозга и периферической нервной системы. Простетической группой тирозингидроксилазы, как и дофамингидроксилазы (последняя катализирует превращение дофамина в норадреналин) является тетрагидробиоптерин (рис.)
Физиологическая роль тирозингидроксилазы чрезвычайно высока, поскольку катализируемая этим ферментом реакция определяет скорость биосинтеза дофамина и катехоламинов, регулирующих в известной степени деятельность сердечно-сосудистой системы. В медицинской практике широко используются, кроме того, ингибиторы декарбоксилазы ароматических аминокислот, в частности α-метилдофа (альдомет), введение которого способствует снижению кровяного давления.

В животных тканях с высокой скоростью протекает реакция декарбоксилирования гистидина, катализируемая специфической гистидиндекарбоксилазой (рис.).
Продукт реакции — гистамин — обладает широким спектром биологического действия. По сосудорасширяющему эффекту на кровеносные сосуды он резко отличается от других биогенных аминов, оказывающих сосудосуживающее действие. Много гистамина образуется в области воспаления, что имеет определенный биологический смысл. Вызывая расширение сосудов в очаге воспаления, гистамин тем самым ускоряет приток лейкоцитов, способствуя борьбе защитных сил организма с инфекцией. Гистамин, кроме того, участвует в секреции НС1 в желудке, что широко используется в клинике при изучении секреторной деятельности желудка (гистаминовая проба). Он имеет прямое отношение к явлениям сенсибилизации и десенсибилизации. При повышенной чувствительности к гистамину в клинике используются антигистаминные препараты (санорин, димедрол и др.), оказывающие влияние на рецепторы сосудов. Гистамину приписывают, кроме того, роль медиатора боли. Болевой синдром, несомненно, является весьма сложным процессом, детали которого пока не выяснены, но участие в нем гистамина не подлежит сомнению.
В клинике широко используются, кроме того, продукт α-декарбоксилирования глутаминовой кислоты — γ-аминомасляная кислота (ГАМК). Фермент, катализирующий эту реакцию (глутаматдекарбоксилаза), является высокоспецифичным:
Интерес к γ-аминомасляной кислоте связан с ее тормозящим действием на деятельность ЦНС. Больше всего γ-аминомасляной кислоты и глутаматдекарбоксилазы обнаружено в сером веществе коры головного мозга, в тo время как белое вещество мозга и периферическая нервная система их почти не содержат. Введение γ-аминомасляной кислоты вызывает разлитой тормозной процесс в коре (центральное торможение) и у животных приводит к утрате условных рефлексов. γ-Аминомасляная кислота используется в клинике при лечении некоторых заболеваний ЦНС, связанных с резким возбуждением коры головного мозга. Так, в практике лечения эпилепсии хороший эффект (резкое сокращение частоты эпилептических припадков) давало введение глутаминовой кислоты. Как оказалось, лечебный эффект был обусловлен не глутаминовой кислотой, а продуктом ее декарбоксилирования γ-аминомаслянной кислотой.
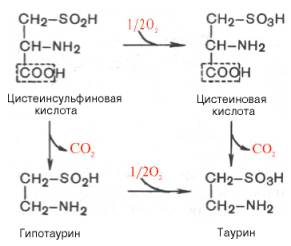
В животных тканях с высокой скоростью декарбоксилируются также два производных цистеина — цистеиновая и цистеинсульфиновая кислоты; в процессе этих специфических ферментативных реакций образуется таурин, который используется в организме для синтеза парных желчных кислот (см. Обмен липидов).

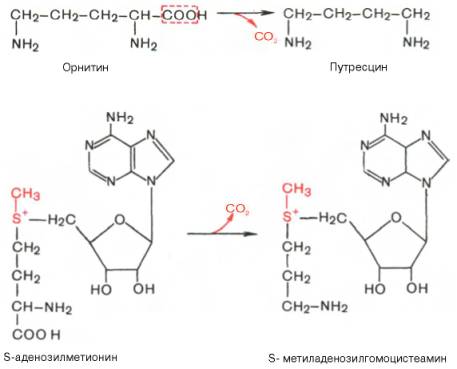
Следует указать еще на два недавно открытых в тканях животных фермента, катализирующих декарбоксилирование орнитина и S-аденозилметионина с образованием путресцина и S-метиладенозилгомоцистеамина:


Значение этих реакций, катализирующихся специфическими орнитиндекарбоксилазой и S-аденозилметиониндекарбоксилазой тканей животных, огромно, если учесть, что путресцин и аминопропильная часть S-метиладенозилгомоцистеамина используются для синтеза полиаминов — спермидина и спермина:
Полиамины, к которым относят также путресцин, оказались необходимыми для регуляции биосинтеза внутриклеточных полимерных молекул (нуклеиновых кислот и белков), хотя конкретная их роль в этом процессе не всегда ясна.
Таким образом, биогенные амины являются сильными фармакологически активными веществами, оказывающими разносторонее влияние на физиологические функции организма. Некоторые биогенные амины нашли широкое применение в качестве лекарственных средств.
Декарбоксилирование аминокислот
Процесс отщепления карбоксильной группы аминокислот в виде СО2 получил название декарбоксилирования. Несмотря на ограниченный круг аминокислот и их производных, подвергающихся декарбоксилиро-ванию в животных тканях, образующиеся продукты реакции – биогенные амины – оказывают сильное фармакологическое действие на множество физиологических функций человека и животных. В животных тканях установлено декарбоксилирование следующих аминокислот и их производных: тирозина, триптофана, 5-окситриптофана, валина, серина, гистидина, глу-таминовой и γ-оксиглутаминовой кислот, 3,4-диоксифенилаланина, цис-теина, аргинина, орнитина, S-аденозилметионина и α-аминомалоновой кислоты. Помимо этого, у микроорганизмов и растений открыто де-карбоксилирование ряда других аминокислот.
1. α-Декарбоксилирование, характерное для тканей животных, при котором от аминокислот отщепляется карбоксильная группа, стоящая по соседству с α-углеродным атомом. Продуктами реакции являются СО2 и биогенные амины:
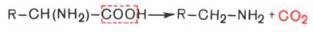
2. ω-Декарбоксилирование, свойственное микроорганизмам. Например, из аспарагиновой кислоты этим путем образуется α-аланин:

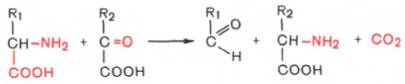
В этой реакции образуются альдегид и новая аминокислота, соответствующая исходной кетокислоте.
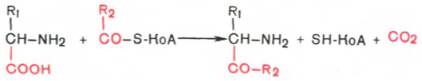
Эта реакция в тканях животных осуществляется при синтезе δ-амино-левулиновой кислоты из глицина и сукцинил-КоА (см. главу 13) и при синтезе сфинголипидов, а также у растений при синтезе биотина.
Реакции декарбоксилирования в отличие от других процессов промежуточного обмена аминокислот являются необратимыми. Они катализируются специфическими ферментами – декарбоксилазами аминокислот, отличающимися от декарбоксилаз α-кетокислот (см. главу 10) как белковым компонентом, так и природой кофермента. Декарбоксилазы аминокислот состоят из белковой части, обеспечивающей специфичность действия, и простетической группы, представленной пиридоксальфосфатом (ПФ), как и у трансаминаз.
Таким образом, в двух совершенно различных процессах обмена аминокислот участвует один и тот же кофермент. Исключение составляют две декарбоксилазы: гистидиндекарбоксилаза Micrococcus и Lactobacilus и аденозилметионин-декарбоксилаза Е. coli, содержащие вместо ПФ остаток пировиноградной кислоты.
Механизм реакции декарбоксилирования аминокислот в соответствии с общей теорией пиридоксалевого катализа (см. рис. 12.3) сводится к образованию ПФ-субстратного комплекса, представленного, как и в реакциях трансаминирования, шиффовым основанием ПФ и аминокислоты:
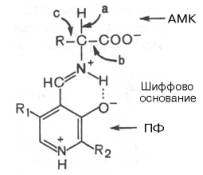
Образование подобного комплекса в сочетании с некоторым оттягиванием электронов белковой частью молекулы фермента сопровождается лабилизацией одной из трех связей при α-углеродном атоме, благодаря чему аминокислота способна вступать в реакции трансаминирования (а), декарбоксилирования (b) и альдольного расщепления (с).
Далее представлены отдельные примеры декарбоксилирования аминокислот, в частности тех, продукты реакции которых оказывают сильное фармакологическое действие. Одним из хорошо изученных ферментов является декарбоксилаза ароматических аминокислот. Она не обладает строгой субстратной специфичностью и катализирует декарбок-силирование L-изомеров триптофана, 5-окситриптофана и 3,4-диоксифе-нилаланина (ДОФА); продуктами реакций, помимо СО2, являются соответственно триптамин, серотонин и диоксифенилэтиламин (дофамин).
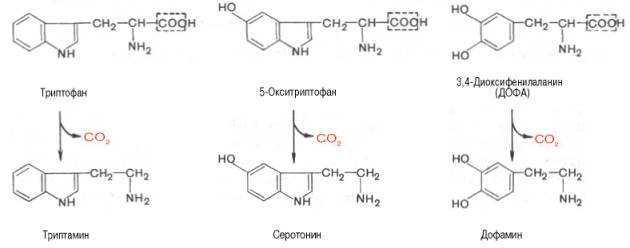
Декарбоксилаза ароматических аминокислот получена в чистом виде (мол. масса 112000), кофермент – ПФ. В больших количествах она содержится в надпочечниках и ЦНС, играет важную роль в регуляции содержания биогенных аминов. Образующийся из 5-окситриптофана серо-тонин оказался высокоактивным биогенным амином сосудосуживающего действия. Серотонин регулирует артериальное давление, температуру тела, дыхание, почечную фильтрацию и является медиатором нервных процессов в ЦНС. Некоторые авторы считают серотонин причастным к развитию аллергии, демпинг-синдрома, токсикоза беременных, карциноидного синдрома и геморрагических диатезов.
Продукт декарбоксилазной реакции дофамин является предшественником катехоламинов (норадреналина и адреналина). Источником ДОФА в организме является тирозин, который под действием специфической гидроксилазы превращается в 3,4-диоксифенилаланин (см. главу 8). Тиро-зин-3-монооксигеназа открыта в надпочечниках, ткани мозга и периферической нервной системы. Простетической группой тирозин-моноокси-геназы, как и дофамин-монооксигеназы (последняя катализирует превращение дофамина в норадреналин) является тетрагидробиоптерин, имеющий следующее строение:
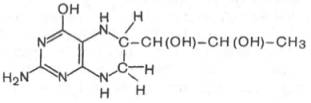
Физиологическая роль тирозин-3-монооксигеназы чрезвычайно велика, поскольку катализируемая этим ферментом реакция определяет скорость биосинтеза катехоламинов, регулирующих деятельность сердечно-сосудистой системы. В медицинской практике широко используются ингибиторы декарбоксилазы ароматических аминокислот, в частности α-метилдофа (альдомет), вызывающий снижение артериального давления.
В животных тканях с высокой скоростью протекает декарбоксилирование гистидина под действием специфической декарбоксилазы.
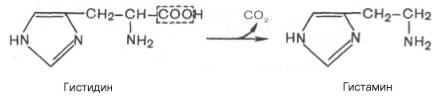
Гистамин оказывает широкий спектр биологического действия. По механизму действия на кровеносные сосуды он резко отличается от других биогенных аминов, так как обладает сосудорасширяющим свойством. Большое количество гистамина образуется в области воспаления, что имеет определенный биологический смысл. Вызывая расширение сосудов в очаге воспаления, гистамин тем самым ускоряет приток лейкоцитов, способствуя активации защитных сил организма. Кроме того, гистамин участвует в секреции соляной кислоты в желудке, что широко используется в клинике при изучении секреторной деятельности желудка (гистаминовая проба). Он имеет прямое отношение к явлениям сенсибилизации и десенсибилизации. При повышенной чувствительности к гистамину в клинике используют антигистаминные препараты (санорин, димедрол и др.), оказывающие влияние на рецепторы сосудов. Гистамину приписывают также роль медиатора боли. Болевой синдром – сложный процесс, детали которого пока не выяснены, но участие в нем гистамина не подлежит сомнению.
В клинической практике широко используется, кроме того, продукт α-декарбоксилирования глутаминовой кислоты – γ-аминомасляная кислота (ГАМК). Фермент, катализирующий эту реакцию (глутаматдекарбоксилаза), является высокоспецифичным.

Интерес к ГАМК объясняется ее тормозящим действием на деятельность ЦНС. Больше всего ГАМК и глутаматдекарбоксилазы обнаружено в сером веществе коры большого мозга, в то время как белое вещество мозга и периферическая нервная система их почти не содержат. Введение ГАМК в организм вызывает разлитой тормозной процесс в коре (центральное торможение) и у животных приводит к утрате условных рефлексов. ГАМК используется в клинике как лекарственное средство при некоторых заболеваниях ЦНС, связанных с резким возбуждением коры большого мозга. Так, при эпилепсии хороший эффект (резкое сокращение частоты эпилептических припадков) дает введение глутаминовой кислоты. Как оказалось, лечебный эффект обусловлен не самой глутаминовой кислотой, а продуктом ее декарбоксилирования – ГАМК.
В животных тканях с высокой скоростью декарбоксилируются также два производных цистеина – цистеиновая и цистеинсульфиновая кислоты. В процессе этих специфических ферментативных реакций образуется таурин, который используется в организме для синтеза парных желчных кислот (см. главу 11).
Следует указать еще на два недавно открытых в тканях животных фермента, катализирующих декарбоксилирование орнитина и S-аденозилметионина: орнитиндекарбоксилазу и аденозилметиониндекарбоксилазу.
Значение этих реакций для тканей животных огромно, поскольку продукты реакций используются для синтеза полиаминов – спермидина и спермина.
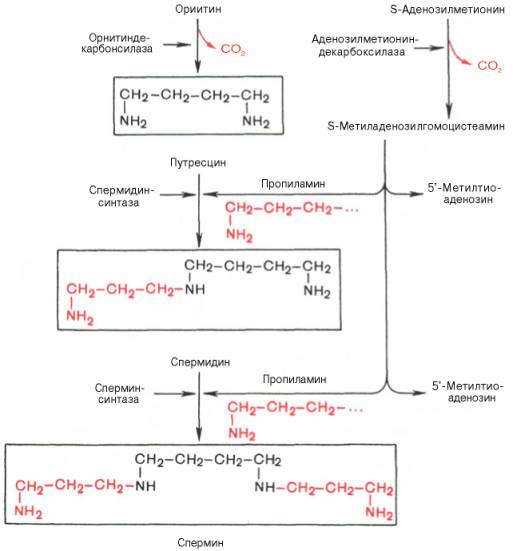
Полиамины, к которым относят также диамин путресцин, играют важную роль в процессах клеточного роста и дифференцировки, в регуляции синтеза ДНК, РНК и белка, стимулируя транскрипцию и трансляцию (см. далее), хотя конкретный механизм участия их в указанных процессах не всегда ясен.
Таким образом, биогенные амины являются сильными фармакологически активными веществами, оказывающими разностороннее влияние на физиологические функции организма. Некоторые биогенные амины нашли широкое применение в качестве лекарственных препаратов.
Распад биогенных аминов. Накопление биогенных аминов может отрицательно сказываться на физиологическом статусе и вызывать ряд существенных нарушений функций в организме. Однако органы и ткани, как и целостный организм, располагают специальными механизмами обезвреживания биогенных аминов, которые в общем виде сводятся к окислительному дезаминированию этих аминов с образованием соответствующих альдегидов и освобождением аммиака:
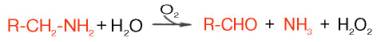
Ферменты, катализирующие эти реакции, получили название моноамин-и диаминоксидаз. Более подробно изучен механизм окислительного дез-аминирования моноаминов. Этот ферментативный процесс является необратимым и протекает в две стадии:
Первая (1), анаэробная, стадия характеризуется образованием альдегида, аммиака и восстановленного фермента. Последний в аэробной фазе окисляется молекулярным кислородом. Образовавшаяся перекись водорода далее распадается на воду и кислород. Моноаминоксидаза (МАО), ФАД-содержащий фермент, преимущественно локализуется в митохондриях, играет исключительно важную роль в организме, регулируя скорость биосинтеза и распада биогенных аминов. Некоторые ингибиторы моно-аминоксидазы (ипраниазид, гармин, паргилин) используются при лечении гипертонической болезни, депрессивных состояний, шизофрении и др.
Лекция № 26 аминокислоты

А М И Н О К И С Л О Т Ы
Аминокислотами называют бифункциональные производные углеводородов, которые содержат карбоксильную группу —COOH и аминогруппу —NH2.
По систематической номенклатуре аминокислоты называют, по соответствующей карбоновой кислоте добавляя приставку амино-. Положение аминогруппы в углеродной цепи указывают цифрой:

Подробнее номенклатурные правила для названий аминокислот изложены в пособии , «Номенклатура, классификация и электронное строение химических связей в органических соединениях», раздел 4.3.
В зависимости от положения аминогруппы по отношению к карбоксильной группе различают α, β, γ и так далее аминокислоты:

 Все природные аминокислоты содержат аминогруппу только в
Все природные аминокислоты содержат аминогруппу только в
α-положении и имеют общую формулу:
Помимо систематической, для природных аминокислот широко распространена тривиальная номенклатура (аланин, валин, лизин и т. д.). Иногда запись аминокислот осуществляют, используя трёх — буквенные сокращения (Ala, Val, Lys и др.).
В настоящее время единой классификации аминокислот не существует.
Аминокислоты делят на природные (содержатся в растительных и животных организмах) и синтетические – получены искусственным путем.
Организм синтезирует аминокислоты главным образом из пищевых белков. Но есть целая группа аминокислот, которых организм сам синтезировать не может. Эти аминокислоты называют незаменимыми. К ним относятся (валин, лейцин, изолейцин, лизин, треонин, метионин, фенилаланин и триптофан) Такие аминокислоты должны поступать в организм извне.
В настоящее время известно свыше 150 аминокислот, но только 20 из них входят в состав белков.
По природе радикала аминокислоты делят на:
Строение радикала кислоты



Строение радикала кислоты



Строение радикала кислоты


4. Аминокислоты, содержащие в радикале дополнительную аминогруппу или гуанидильный остаток.
Строение радикала кислоты


Аргинин (содержит гунидиновую группу)
5. Аминокислоты, которые содержат в радикале дополнительную карбоксильную или амидную группы:
Строение радикала кислоты




6. Ароматические и гетероциклические аминокислоты:
Строение радикала кислоты




Пролин (полная форма)
Современная рациональная классификация основана на полярности радикалов. Полярность радикала во многом определяет такое важное свойство аминокислот как растворимость в воде и в других полярных растворителях. Полярные группы радикала (-COOH, — NH2, — OH и др.) притягивают воду и тем самым повышают растворимость аминокислот в воде, неполярные радикалы, наоборот, отталкивают воду и снижают растворимость аминокислот в воде.
Все природные α-аминокислоты, кроме глицина (NH2 — CH2 — COOH), имеют асимметрический атом углерода (α-углеродный атом), а некоторые из них даже два хиральных центра, например, треонин. Таким образом, все аминокислоты могут существовать в виде пары несовместимых зеркальных антиподов (энантиомеров).
За исходное соединение, с которым принято сравнивать строение
α-аминокислот, условно принимают D — и L-молочные кислоты, конфигурации которых, в свою очередь, установлены по D — и L-глицериновым альдегидам.


Все превращения, которые осуществляются в этих рядах при переходе от глицеринового альдегида к α-аминокислоте, выполняются в соответствии с главным требованием — они не создают новых и не разрывают старых связей у асимметрического центра.
Для определения конфигурации α-аминокислоты в качестве эталона часто используют серин (иногда аланин). Конфигурации их так же выведены из D — и L-глицериновых альдегидов:

Природные аминокислоты, входящие в состав белков, относятся к L-ряду.
D-формы аминокислот встречаются сравнительно редко, они синтезируются только микроорганизмами и называются «неприродными» аминокислотами. Животными организмами D-аминокислоты не усваиваются. Интересно отметить действие D — и L-аминокислот на вкусовые рецепторы: большинство аминокислот L-ряда имеют сладкий вкус, а аминокислоты D-ряда — горькие или безвкусные.
Без участия ферментов самопроизвольный переход L-изомеров в D-изомеры с образованием эквимолярной смеси (рацемическая смесь) осуществляется в течение достаточно длительного промежутка времени.
Рацемизация каждой L-кислоты при данной температуре идет с определенной скоростью. Это обстоятельство можно использовать для установления возраста людей и животных. Так, например, в твердой эмали зубов имеется белок дентин, в котором L-аспартат переходит в D-изомер при температуре тела человека со скоростью 0,01% в год. В период формирования зубов в дентине содержится только L-изомер, поэтому по содержанию D-аспартата можно рассчитать возраст человека или животного.
Физические свойства аминокислот
Хотя аминокислоты обычно изображают как соединения, содержащие амино — и карбоксильную группы (H2N — CHR — COOH), некоторые их свойства, как физические, так и химические, не согласуются с этой структурой. Присутствие в молекуле у одного атома углерода двух функциональных групп приводит к появлению ряда специфических свойств.
Во-первых, в противоположность аминам и карбоновым кислотам аминокислоты представляют собой нелетучие кристаллические вещества, плавящиеся с разложением при близких и довольно высоких температурах, поэтому идентификации аминокислот по температурам плавления достаточно затруднительна.
Во-вторых, аминокислоты очень плохо растворимы в неполярных растворителях типа петролейного эфира, диэтилового эфира, бензола и хорошо растворимы в воде.
В-третьих, в водных растворах аминокислоты имеют высокие дипольные моменты.
В-четвертых, константы кислотности и основности для групп СООН и NH2 необычайно малы. Так, для глицина константа кислотности Ka = 1,6×10-10, а константа основности Kb = 2,5×10-12; в то время как для большинства карбоновых кислот Ka » 10-5 а для алифатических аминов Kb » 10-4. Все эти свойства вполне объяснимы, если принять во внимание тот факт, что аминокислоты существуют в виде диполярного иона, который образуется за счет отщепления протона от карбоксильной группы и присоединения его к аминогруппе. Диполярный ион часто называют внутренней солью.

Кислотно-основные свойства также становятся понятными, если учесть, что измеряемая Ka в действительности относится к кислотности иона RNH3+:

а константа основности (Kb) в действительности относится к основности карбоксилат-иона.

При подщелачивании раствора аминокислоты диполярный ион I превращается в анион II, так как более сильное основание (гидроксильный ион) отрывает протон от иона аммония и образуется более слабое основание — амин.

Если подкислить раствор аминокислоты, ион I превратится в катион III, так как более сильная кислота Н3О+ отдает протон карбоксилат-иону и образуется более слабая кислота:

Необходимо отметить, что ионы II и Ш, содержащие свободную аминогруппу или свободную карбоксильную группу, находятся в равновесии с диполярным ионом:

Однако следует иметь в виду, что в данном равновесии участвует также определенное (хотя и небольшое) количество незаряженных молекул аминокислот.
Изоэлектрическая точка аминокислот
Мы рассмотрели превращение в кислой и щелочной средах моноаминомонокарбоновых кислот, в радикалах которых не содержится ионогенных групп (аминокислоты с недиссоциирующими радикалами).
Изменение суммарного заряда аминокислот с анионными и катионными группами в радикале, в зависимости от рН среды, можно представить в следующей таблице. Для сравнения в эту же таблицу поместим аминокислоты, в радикале которых нет диссоциирующих групп.
В сильнокислом растворе имеется значительный избыток катионов, а в сильно щелочном — избыток анионов.
Если раствор аминокислоты поместить в электрическое поле, то в зависимости от активной реакции среды будет наблюдаться следующая картина: в кислой среде ион аминокислоты мигрирует к катоду, а в щелочной — к аноду. Если при определенном рН среды концентрация катионов станет равной концентрации анионов, то никакого движения аминокислоты происходить не будет.
Концентрация ионов водорода (pH), при которой аминокислота не перемещается в электрическом поле, называется изоэлектрической точкой данной аминокислоты (рI).
Изоэлектрическая точка аминокислоты зависит от кислотности группы — NH3+, основности карбоксилат-аниона, природы радикала и присутствия в молекуле кислоты любой дополнительной основной или кислотной группы.
При pH ≠ pI в растворе присутствует равновесная смесь диполярного иона и катионной или анионной формы, что в некоторых случаях может привести к появлению у растворов аминокислот буферных свойств (подробнее см. учебное пособие «Общая химия, часть III» под редакцией профессора , глава «Буферные системы»). Значительной буферной ёмкостью в интервале физиологических значений рН, (т. е. в интервале 6-8) обладает только гистидин. Отметим лишь, что при pH = pI растворы аминокислот буферного действия не проявляют.
При пропускании постоянного тока через раствор, содержащий смесь нескольких аминокислот, каждая из них будет двигаться к катоду или к аноду со скоростью, зависящей от природы этой аминокислоты и от рН среды. Разделение и анализ смесей аминокислот, основанное на этом явлении, называется электрофорезом.
Химические свойства аминокислот
Наличие в молекуле аминокислоты функциональных групп кислотного и основного характера обусловливает амфотерность аминокислот. Подобно любому амфотерному соединению, аминокислоты образуют соли как при действии кислоты, так и при действии щелочи.

Аминокислоты, будучи гетерофункциональными соединениями, должны проявлять свойства как одной, так и другой функциональной группы.
Реакции карбоксильной группы
1. Образование внутрикомплексных солей.
С катионами тяжелых металлов α-аминокислоты образуют внутрикомплексные соли. Так, со свежеприготовленным гидроксидом меди (II) α-аминокислоты образуют хорошо кристаллизующиеся хелатные соли меди (II), окрашенные в синий цвет:

2. Образование сложных эфиров.
Так как реакция этерификации протекает в кислой среде, сложные эфиры аминокислот образуются в виде солей по аминогруппе:

Образовавшиеся эфиры не могут существовать в виде биполярных ионов, поэтому, в отличие от исходных аминокислот, они растворяются в органических растворителях и имеют более низкие температуры кипения. Это даёт возможность разделить смесь эфиров аминокислот перегонкой.
3. Образование хлорангидридов.
Эту реакцию часто называют реакцией «активации» карбоксильной группы. Хлорангидриды α-аминокислот получают действием на аминокислоты тионилхлорида (SOCl2) или хлорида фосфора (V) (PCl5). Полученные хлорангидриды неустойчивы и существуют только в виде солей:

Поэтому реакцию обычно проводят, предварительно защитив аминогруппу ацилированием.
4. Образование амидов аминокислот.
Такие амиды получают действием аммиака или первичных аминов на хлорангидриды с защищённой аминогруппой. В случае использования реакции с аминами получают замещённые по азоту амиды аминокислот:

5. Декарбоксилирование аминокислот.
В лабораторных условиях эта реакция протекает при нагревании аминокислоты с Ba(OH)2. В результате получается первичный амин:

Все реакции карбоксильной группы аминокислот можно представить следующей схемой:

1. Реакция ацилирования. Образование N-замещённых амидов.
N-замещенные амиды часто рассматривают как N-ацильные производные. Эта реакция была отмечена ранее как реакция защиты аминогруппы. Её можно рассматривать как процесс ацилирования аминогруппы хлорангидридами или ангидридами кислот:

Реакция протекает лучше в щелочной среде. Примером может служить получение N-бензоилаланина в присутствии водного раствора гидроксида натрия. Этот метод получения N-ацильных производных называют ацилированием по Шоттен-Бауману:

Щёлочь необходима для связывания выделяющегося хлороводорода, т. к. в кислой среде N-ацильные производные легко гидролизуются, освобождая исходную аминокислоту:

Это общепринятый способ удаления защитной группы. Однако в некоторых случаях невозможно удалять защитную группу гидролизом в кислой среде. Например, при гидролизе пептидов будет разрушаться пептидная связь. В этих случаях защиту проводят такими реагентами, удаление которых можно провести не гидролизом, а каким-либо другим методом. Например, аминогруппу можно защищать реакцией с карбобензоксихлоридом (бензиловый эфир хлормуравьиной кислоты). Карбобензоксигруппа удаляется затем каталитическим гидрогенолизом:

2. Алкилирование аминокислот.
Аминокислоты можно алкилировать по аминогруппе галоидными алкилами (обычно иодистыми алкилами). Например, алкилированием глицина можно получить метиламиноуксусную кислоту — саркозин, которая в связанном виде содержится в некоторых белках.

При избытке иодистого метила образуется четвертичная аммонийная соль:

Реакция протекает так же, как и при взаимодействии с азотистой кислотой алифатических первичных аминов — выделяется азот, а аминогруппа замещается на гидроксильную группу:

Таким образом можно установить структурное родство аминокислот с соответствующими оксикислотами. По объёму выделившегося азота определяют количество α-аминокислоты, вступившей в реакцию (метод Ван-Слайка).
4. Взаимодействие с альдегидами.
α-Аминокислоты, подобно первичным аминам, реагируют с альдегидами, образуя замещенные имины (основания Шиффа). Реакция протекает через стадию образования карбиноламинов.

При взаимодействии α-аминокислот с формальдегидом образуются относительно устойчивые карбиноламины — N-метилольные производные, свободная карбоксильная группа которых может быть оттитрована щелочью.
Формальдегид, взятый в избытке, способствует отщеплению протона от NH3+ группы биполярного иона и легко соединяется со свободной (непротонированной) аминогруппой, образуя устойчивое метилольное производное.

Титрование аминокислоты в избытке формальдегида (формольное титрование) представляет собой аналитический метод (метод Серенсена), при помощи которого прослеживается, в частности, образование свободных аминокислот в процессе гидролиза белков.
5. Взаимодейстивие с динитрофторбензолом (ДНФБ).
Важной реакцией α-аминогруппы является её реакция с
2,4-динитрофторбензолом (ДНФБ) в слабощелочном растворе, которую впервые использовал Фредерик Сенгер для количественного введения метки в аминогруппы аминокислот и пептидов. Эта реакция протекает по механизму нуклеофильного замещения.

Продукт реакции окрашен в интенсивно желтый цвет. Эта реакция представляет исключительную ценность для идентификации N-концевых аминокислот полипептидных цепей.
Все вышеперечисленные реакции аминогруппы аминокислот можно представить следующей схемой:

Реакции функциональных групп, содержащихся в радикалах аминокислот
Аминокислоты вступают также в реакции, типичные для функциональных групп, присутствующих в их радикалах. Например, для SH-групп цистеина, гидроксильной группы тирозина и треонина, гуанидиновой группы аргинина.
1. Реакции сульфгидрильной (тиоловой) группы.
Для сульфгидрильной группы характерна исключительно высокая реакционная способность. Например, при действии на цистеин незначительных концентраций ионов некоторых тяжелых металлов образуются меркаптиды.

В щелочных растворах цистеин легко теряет атом серы. Так, при нагревании цистеина с ацетатом свинца в щелочном растворе образуется черный осадок сульфида свинца. Эта реакция применяется для обнаружения сульфгидрильной группы в пептидах и белках.
Тиоловая группа цистеина легко подвергается окислению с образованием дисульфида. Этот процесс можно отразить следующей схемой:

Дисульфидные связи, присоединяя два атома водорода, переходят в сульфгидрильные (тиоловые) группы:

Рассмотрим этот процесс на примере превращения цистеина в цистин:

В цистине при действии восстановителей дисульфидная связь разрывается и образуется две молекулы цистеина:

Дисульфидная связь может также подвергаться окислению под действием таких жестких окислителей, как, например, надмуравьиная кислота. В результате образуется цистеиновая кислота:

2. Реакции гидроксильной группы – реакции элиминирования.
Эти реакции характерны для аминокислот, содержащих в радикале гидроксильную группу в β-положении по отношению к карбоксильной группе (серин и треонин).
В результате ряда последовательных реакций аминокислота превращается в кетокислоту. Рассмотрим этот процесс на примере превращения треонина в 2-оксобутановую кислоту.

3. Реакции гуанидильной группы.
Гуанидильная группа содержится в радикале аргинина:

Гуанидильная группа аргинина легко отщепляется при гидролизе в избытке гидроксида бария при 1000С с образованием мочевины и орнитина:

Орнитин — α-аминокислота, содержащая в радикале вторую аминогруппу, в состав белков не входит. Появляется в организме в результате гидролитического расщепления аргинина с участием фермента аргиназы. Аргиназа в значительных количествах содержится в печени и в малых количествах в почках и селезенке млекопитающих животных.
Специфические реакции α-аминокислот
Присутствие у одного атома углерода двух функциональных групп (аминогруппы и карбоксильной) приводит к появлению специфических реакций.
1. Образование пептидов — реакция ацилирования одной аминокислоты другой аминокислотой:

Затем дипептид присоединяет следующую молекулу аминокислоты, образуя трипептид, и так далее:

2. Межмолекулярная циклизация — образование дикетопиперазинов.
При отщеплении двух молекул воды от двух молекул аминокислот образуется циклический дипептид — дикетопиперазин:

Реакции аминокислот in vivo
Простые аминокислоты, как и многие другие простые «биологические молекулы», не накапливаются в клетке: как правило, их избыток разрушается при помощи реакций, которые снабжают живую систему энергией. Три основные реакции, катализируемые ферментами, благодаря которым осуществляется превращение аминокислот в клетке, это реакции дезаминирования, переаминирования и декарбоксилирования.
1. Дезаминирование аминокислот
В организме дезаминирование может осуществляться как неокислительным, так и окислительным путём.
Неокислительное дезаминирование встречается, в основном, у бактерий и грибов. Например, превращение аспарагиновой кислоты в фумаровую под действием фермента аспартазы.

Окислительное дезаминирование — протекает при участии фермента оксидазы. Для того чтобы полностью прошла реакция окислительного дезаминирования, фермент, катализирующий эту реакцию, нуждается в окислительном (дегидрирующем) агенте. Обычно акцептором водорода в таких системах служит ФАД (флавинадениндинуклеотид), который затем переходит в восстановленную форму, сокращённо обозначаемую ФАД-Н2.
Окислительное дезаминирование осуществляется через стадию образования промежуточного имина.
Рассмотрим процесс превращения аланина в пировиноградную кислоту.

Реакции дезаминирования позволяют организму удалять избыток аминокислот, однако при этом повышается концентрация нежелательных азотистых веществ. Высокие концентрации аммиака и его производных токсичны для организма, который поэтому стремится освободиться от них, выделяя лишний азот в виде мочевины или мочевой кислоты.

Мочевая кислота образуется в организме взрослого человека в качестве побочного продукта. Высокое содержание мочевой кислоты приводит к мочекаменной болезни. Мочевая кислота в виде кристаллов мононатриевой соли образует камни в почках и в мочевом пузыре. Соли мочевой кислоты в суставах вызывают болезненные симптомы подагры — очень широко распространенного заболевания человека. Содержание мочевой кислоты и её солей в организме человека может представлять интерес с точки зрения эволюционной теории, поскольку большинство животных полностью разлагают мочевую кислоту до её выделения из организма. Было высказано предположение о том, что присутствие мочевой кислоты в организме человека предоставляет людям некоторое эволюционное преимущество. Эта гипотеза ещё не доказана, но она может быть интересным связующим звеном между биохимическими свойствами вещества и поведением живых организмов.
2. Переаминирование (трансамнирование).
Реакция сводится к взаимопревращению аминогруппы и карбонильной группы под действием ферментов трансаминаз.
Эта реакция служит не только для разрушения аминокислот, но и для их биосинтеза. Рассмотрим реакцию взаимопревращения аспарагиновой кислоты и α-кетоглутаровой в щавелевоуксусную и глутаминовую кислоты:

Эта схема не отражает истинного механизма процесса.
Данное взаимопревращение нуждается в пиридоксальфосфате, который образует имин с исходной аминокислотой, сохраняет аминогруппу при превращении аминокислоты в соответствующую α-кетокислоту и образует имин с другой α-кетокислотой.
Рассмотрим процесс превращения аминокислоты I в α-кетокислоту I и
α-кетокислоты II в аминокислоту II.
Альдегидная группа пиридоксальфосфата образует имин с аминокислотой I, имин далее изомеризуется и после гидролиза выделяет кетокислоту I и пиридоксаминфосфат.

Таким образом, из исходной аминокислоты получилась кетокислота. Образовавшийся пиридоксаминфосфат далее реагирует с другой кетокислотой (кетокислота II), образуя имин, содержащий радикал новой кетокислоты (R¢). Имин далее изомеризуется и после гидролиза образует новую аминокислоту (аминокислота II):

По завершении всей сложной последовательности реакций, после гидролиза пиридоксальфосфат регенерируется и способен принять участие в следующих взаимопревращениях аминокислот и α-кетокислот.
Как своеобразную реакцию взаимопревращения аминокислоты и амидоаминокислоты, сопровождающуюся заменой амидогруппы одной аминокислоты на гидроксильную группу другой, можно рассматривать реакцию взаимодействия L-аспарагиновой кислоты и L-глутамина, катализируемую аспарагинсинтетазой в присутствии АТФ, и приводящую к образованию
L-аспарагина и L-глутаминовой кислоты.

3. Декарбоксилирование аминокислот.
Декарбоксилирование in vivo — это путь образования биогенных аминов. В организме эта реакция катализируется ферментами — декарбоксилазами. Некоторые амины обладают ярко выраженной биологической активностью. Интересной, например, является реакция образование дофамина при декарбоксилировании диоксифенилаланина, поскольку дофамин — это биологический предшественник адреналина.

В реакции декарбоксилирования, которая протекает при гниении белков, лизин и орнитин, образуют диамины: кадаверин и путресцин.

Интересной является реакция декарбоксилирования глутаминовой кислоты, так как она приводит к образованию γ-аминомасляной кислоты, которую рассматривают как природный транквилизатор.
Этот процесс также нуждается в присутствии пиридоксальфосфата.

Ярко выраженной биологической активностью обладает амин, образующийся при декарбоксилировании гистидина:

Гистамин является медиатором аллергии: он расширяет все периферические сосуды, что приводит к резкому падению артериального давления, нарушает проницаемость сосудистой стенки, что может быть одной из причин появления отеков, вызывает бронхоспазм и. т.д. Группа препаратов, применяемых в медицине для уменьшения проявления аллергических реакций, так или иначе связанных с гистамином, была названа антигистаминными препаратами.
4. Реакции гидроксилирования и карбоксилирования.
С помощью этих реакций в молекулу органического соединения вводится дополнительная гидроксильная или карбоксильная группы. Реакции протекают при участии соответствующих ферментов и приводят к образованию модифицированных аминокислот. Эти реакции не имеют аналогов в химии in vitro.
Гидроксилированием называют введение в молекулу органического соединения гидроксильной группы. Так, гидроксилирование фенилаланина приводит к образованию тирозина:

Отсутствие в организме фермента, катализирующего эту реакцию, приводит к тяжелому заболеванию — фенилкетонурии.
Значительный интерес представляет реакция гидроксилирования пролина:

Гидроксилирование пролина необходимо для стабилизации тройной спирали коллагена, которая осуществляется за счет образования водородных связей.
При цинге нарушается гидроксилирование остатков пролина и лизина. В результате образуются менее прочные коллагеновые волокна, что приводит к хрупкости и ломкости кровеносных сосудов.
Карбоксилированием называют введение в молекулу органического соединения карбоксильной группы. Таким образом получают, например,
γ-карбоксиглутаминовую кислоту:

γ-Карбоксиглутаминовая кислота входит в состав белков, участвующих в процессах свертывания крови, так как две близлежащие карбоксильные группы в её структуре способствуют более полному связыванию белковых факторов с ионами кальция:

Нарушение карбоксилирования глутамата приводит к снижению свертываемости крови.
Таким образом, модифицированные аминокислоты, имеющие в своих структурах дополнительные функциональные группы, приобретают свойства, необходимые для выполнения ими специфических функций.
5. Восстановительное аминирование.
Это реакция превращения α-кетокислот в α-аминокислоты осуществляется в организме при участии восстановленной формы никотинамидадениндинуклеотида (НАД∙Н). Так, продуктом метаболизма углеводов является α-кетоглутаровая кислота, которая в результате ряда реакций превращается в глутаминовую кислоту:

6. Альдольное расщепление.
Реакция протекает с α-аминокислотами, содержащими гидроксильную группу в β-положении углеводородного радикала.
Рассмотрим, например, реакцию расщепления серина, в результате которой образуются глицин и формальдегид.

В результате этой реакции расщепляется С-С связь между α- и
β-углеродными атомами. Образующийся формальдегид не выделяется, а связывается с другим коферментом — тетрагидрофолиевой кислотой и в качестве одноуглеродного фрагмента участвует далее в синтезе многих важных соединений.
Полипептиды образуются в результате реакции конденсации, протекающей между аминогруппой одной кислоты и карбоксильной группой другой:

Пептид, образованный двумя аминокислотами, называется дипептид, тремя — трипептид и. т.д. Количество аминокислот в составе пептидов может сильно варьировать. Пептиды, содержащие до 10 аминокислотных остатков, называют олигопептидами. Часто в названии таких молекул указывают число аминокислот, входящих в состав данного олигопетида: дипептид, трипептид, тетрапептид, октапептид и. т.д.
Пептиды, содержащие более 10 аминокислот, называют полипептидами. А полипептиды, содержащие более 50 аминокислотных остатков, обычно называют белками. Однако такие градации весьма условны: например, гормон глюкагон, состоящий из 29 аминокислот, называют белковым гормоном. Гормоны окситоцин и вазопрессин содержат всего по 9 аминокислотных остатков.
Поэтому более удачным следует считать различие, проводимое на уровне структуры полимера, более сложном, чем простая аминокислотная последовательность и количественный состав пептида. Полипептиды представляют собой линейные, довольно гибкие молекулы, а длинные цепи белков свернуты в клубок или иную структуру. Многие белки могут иметь в своем составе группы небелкового характера (простетические группы), связанные с полиамидной цепью.
Пептиды различаются по аминокислотному составу, количеству и порядку соединения аминокислот. Например, тетрапептиды сер-гис-про-ала и ала-гис-про-сер — это два разных пептида, несмотря на то, что они имеют одинаковый качественный и количественный состав.
Строение полипептидной цепи и пептидной связи
Мономеры аминокислот, входящие в состав полипептидов, называют аминокислотными остатками. Аминокислотный остаток, имеющий свободную аминогруппу, называют N-концевым и записывают слева пептидной цепи, а имеющий свободную α-карбо-ксильную группу – С-концевым, и записывают справа. Цепь повторяющихся атомов –СН – СО – NH– в полипетидной цепи называется пептидным остовом.
Полипептидная цепь имеет следующий общий вид:

где R1, R2, R3, … Rn – радикалы аминокислот, образующие боковую цепь.
При названии полипептида к названию всех аминокислотных остатков, кроме последнего, добавляют суффикс —ил, концевая аминокислота имеет окончание —ин. Например, пептид мет-асп-вал-про имеет полное название метиониласпарагилвалилпролин.
Кислотно-основные свойства пептидов
Многие короткие пептиды были получены в чистом кристаллическом виде. Высокие температуры их плавления указывают на то, что из нейтральных растворов пептиды кристаллизуются в виде диполярных ионов. Поскольку ни одна из
α-карбоксильных групп и ни одна из α-аминогрупп, участвующих в образовании пептидных связей, не может ионизироваться в интервале рН от 0 до 14, кислотно-основные свойства пептидов определяются свободной NH2 группой N-концевого остатка и свободной карбоксильной группой С-концевого остатка пептида и теми
R-группами, которые способны к ионизации. В длинных пептидных цепях число ионизированных
R-групп обычно велико по сравнению с двумя ионизированными группами концевых остатков пептида. Поэтому для характеристики кислотно-основных свойств пептидов мы будем рассматривать короткие пептиды.
Свободная α-аминогруппа и свободная концевая карбоксильная группа в пептидах разделены значительно большим расстоянием, чем в простых аминокислотах, и поэтому электростатические взаимодействия между ними ослаблены. Величины рK для концевых карбоксильных групп в пептидах несколько выше, а для концевых α-аминогрупп несколько ниже, чем в соответствующих свободных аминокислотах. У R-групп в коротких пептидах и в соответствующих свободных аминокислотах величины рK заметно не различаются.
Для определения области рН, в которой может находиться изоэлектрическая точка исследуемого короткого пептида, достаточно сравнить число свободных аминогрупп и число свободных карбоксильных групп, включая N — и С-концевые группы. Если число аминогрупп превышает число карбоксильных групп, изоэлектрическая точка пептида будет лежать в щелочной области рН, так как для предотвращения протонирования аминогрупп необходима щелочь. Если число карбоксильных групп превышает число аминогрупп, изоэлектрическая точка будет находиться в кислой области рН, так как кислая среда подавляет диссоциацию карбоксильных групп.
Определение структуры пептидов
Для того чтобы выяснить структуру пептида, необходимо знать следующее:
а) какие аминокислоты входят в состав полипептида;
б) сколько аминокислот каждого вида содержится в пептиде;
в) в какой последовательности эти аминокислоты связаны в цепи.
Для определения состава пептида его подвергают гидролизу в горячей соляной кислоте с С(HCl) = 6 моль/л. Полученную смесь аминокислот анализируют на аминокислотном анализаторе и устанавливают качественный и количественный состав пептида. Зная весовое содержание каждой из полученных аминокислот, можно вычислить количество каждой кислоты и тем самым установить «эмпирическую формулу» пептида, т. е. относительное содержание остатков различных аминокислот в пептиде.
Для вычисления «молекулярной» формулы пептида, то есть для установления действительного числа каждого из остатков в молекуле пептида, необходимо знать его молярную массу, которую определяют различными химическими или физическими методами.
Наиболее трудная задача — установить, в какой последовательности аминокислотные остатки связаны в пептид. Для решения этого вопроса используют комбинацию двух методов: определение концевых групп и частичный гидролиз.
Идентификацию аминокислотных остатков на концах пептидной цепи проводят, используя их отличие от всех остальных звеньев и друг от друга:
N-концевой остаток содержит свободную аминогруппу, а С-концевой остаток содержит свободную карбоксильную группу.
Для идентификации N-концевого остатка используют метод Ф. Сенгера, который основан на реакции свободной аминогруппы пептида с динитрофторбензолом. Реакция протекает по механизму нуклеофильного замещения:

Замещенный пептид подвергают гидролизу, после чего
N-концевой остаток, меченный динитрофенильной группой, выделяют и идентифицируют. N-концевая аминокислота с динитрофторбензолом дает устойчивое, окрашенное в желтый цвет, соединение, которое не разрушается при гидролизе.
Огромный шаг вперед в химии анализа полипептидов был сделан в 1956 году, когда П. Эдман установил, что N-концевую аминокислоту можно удалить при помощи фенилизотиоцианата: (С6Н5 – N = C = S). В результате следующая за ней аминокислота становится N-концевой и её, в свою очередь, также можно удалить, действуя фенилизотиоцианатом. Этот метод определения N-концевых остатков получил название «метод деградации по Эдману».
Наиболее успешным методом определения С-концевых остатков является не химический метод, а ферментативный. Избирательное удаление С-концевого звена осуществляется при помощи фермента карбоксипептидазы, которая расщепляет лишь ту пептидную связь, которая находится в α-положении к свободной
α-карбоксильной группе в полипептидной цепи. Анализ можно повторить на укороченном пептиде, чтобы определить новую С-концевую кислоту.
Однако на практике невозможно определить последовательность остатков аминокислот в длинной пептидной цепи путем ступенчатого удаления концевых остатков. Вместо этого пептид подвергают частичному гидролизу, при котором образуются фрагменты пептидов с укороченной цепью. Эти фрагменты идентифицируют при помощи метода определения концевых групп.
Структура, приписанная пептиду и определенная вышеописанным методом, окончательно подтверждается синтезом этого пептида.