| Название | Кинетика топохимических уравнений. Уравнение Ерофеева, уравнение РогинскогоШульца Уравнение сжимающейся сферы |
| Анкор | Топохимические реакции |
| Дата | 12.09.2020 |
| Размер | 77.64 Kb. |
| Формат файла |  |
| Имя файла | Топохимические реакции.docx |
| Тип | Закон #137621 |
| Подборка по базе: Решение системы линейных уравнений.docx, Дифференциальное уравнение высших порядков.docx, Д 1. Химиялық кинетика_2021.pptx, 14 ноября. Лекция 9. Химическая кинетика и равновесие.pdf, Курсовая. Трипузова К.Д. 2 курс, МО. Приближенное решение алгебр, ОКОНЧАТЕЛЬНЫЙ Уравнение высоты.pdf, ИДЗ 2. Термохимические расчеты и химическая кинетика.pdf, Графический способ решения систем уравнений..ppt, Решение задач Волкенштейн 1.24 Зависимость пройденного телом пут, Метод изоклин Дифференциальное уравнение первого порядка.docx Содержание СодержаниеКинетические закономерности топохимических реакций 3 Образование зародышей 5 Процессы лимитируемые скоростью образования зародышей 9 Гетерогенные процессы 11 Список литературы 17 Кинетические закономерности топохимических реакцийТопохимические реакции – химические реакции, протекающие на поверхности раздела фаз исходного твердого реагента и образующегося твердого продукта. В них могут участвовать и жидкие, и газообразные фазы. Для таких реакций характерно возникновение границы раздела твердых фаз в результате самой реакции. Типичными топохимическими реакциями являются: разложение солей с образованием твердых оксидов, окисление металлов, дегидратация кристаллогидратов. Основные черты топохимической реакции определяются тем, что реагирующие молекулы, атомы или ионы, образующие кристаллическое вещество, жестко закреплены в кристаллических решетках и лишены той подвижности, которой они обладают в газовой или жидкой фазах. Реакционная способность атомов или ионов в значительной степени зависит от того, в каком месте кристалла они находятся – в объеме, на поверхности грани, на ребре кристалла или на вершине. Кроме того, для топохимической реакции большое значение имеет реальная структура твердого тела, и особенно, наличие дефектов решетки. Очень часто при топохимической реакции продукт реакции сохраняет внешнюю форму кристаллов исходного вещества, вследствие того, что при топохимической реакции возможно лишь минимальное перемещение реагирующих частиц. Топохимическая реакция характеризуется специфическими кинетическими закономерностями. Прежде всего, надо отметить своеобразный характер изменения скорости реакции во времени. В начале реакции ее скорость мала (индукционный период), затем скорость возрастает, достигает максимума и снижается либо до нуля, либо до сравнительно малых значений, и слабо зависит от времени. Интегральная кривая, зависимость степени превращения от времени, представляет собой S-образную (сигмоидную) кривую (рис. 1). Скорость реакции находят как тангенс угла наклона касательной к интегральной кривой при заданном времени(d/d), в результате получают дифференциальную кривую топохимической реакции, зависимость скорости реакции от времени (рис. 2). Используют также понятие эмпирической скорости реакции, которая представляет собой максимальную скорость, соответствующую точке перегиба сигмоидной кривой. Отрезок, отсекаемый на оси времени касательной в точке перегиба интегральной кривой называется периодом индукции. Механизм топохимической реакции включает в себя несколько стадий:
Реакционная поверхность представляет собой поверхность раздела фаз исходного вещества и продукта и называется реакционной зоной или фронтом реакции, ее толщина составляет одну молекулу. Таким образом, ходу кривой скорость-время можно дать следующее объяснение. По мере образования новой фазы твердого продукта в реакции появляется поверхность раздела фаз. Размеры ядер растут, повышается и наблюдаемая скорость реакции. Далее растущие ядра фазы нового продукта начинают сливаться и наблюдаемая скорость реакции проходит через максимум. Продолжающееся слияние отдельных ядер приводит к уменьшению поверхности раздела твердых фаз и скорости реакции. После того, как образуется сплошной слой твердого продукта скорость реакции и по мере уменьшения доли непрреагировавшего вещества реакция прекращается. Образование зародышейДля образования зародыша необходима энергия, которая будет затрачена на перестройку кристаллической решетки и образование межфазовой поверхности. Образование новой фазы (зародыша) внутри исходной материнской фазы относится к фазовым переходам первого рода, которые характеризуются скачкообразным изменением объема, энтальпии и энтропии. На поверхности кристалла S0 имеется z0 потенциальных центров, на которых могут возникнуть ядра нового продукта N. Концентрация таких центров может меняться только за счет образования из них ядер. Тогда скорость образования ядер dN/d можно записать: Wуд — удельная скорость процесса образования ядер на 1 центр, N0/S0 – уменьшение числа потенциально возможных центров. Проинтегрируем уравнение и разделив переменные получим Подставив значение N из (2) в (1) и преобразовав, получим Уравнение (3) называют экспоненциальным законом скорости топохимической реакции. Коэффициент Wуд – константой скорости образования зародышей. На ранних стадиях процесса топохимическая реакция хорошо описывается экспоненциальным законом, кроме того, закон справедлив при изотермическом течении процесса и неизменном составе газовой среды. Для описания стадии роста зародышей и их перекрывания применяют множество уравнений, выбор модели описывающей процесс определяется видом кинетической кривой, выражающей экспериментальные данные, а также формой частиц.
степенной закон 1/n = k, экспоненциальный закон ln = k. 2. Уравнения сигмоидного типа: 3. Уравнения кинетики, характеризующиеся диффузионным механизмом: одномерная диффузия 2 = k, двухмерная диффузия (1-)ln(1-) + = k, трехмерная диффузия [1-(1-) 1/3 ] 2 = k, уравнение сжимающейся площади 1-(1-) 1/2 = k, уравнение сжимающегося объема 1-(1-) 1/3 = k, уравнение Гистлинга-Браунштейна [1- 2 /3]-(1-) 2/3 = k. Особенность топохимической реакции состоит в том, что она не только локализована на границе раздела фаз, но и в том, что сама граница с течением процесса сама претерпевает изменение, как своей структуры, так и свойств. Топохимическая реакция является сложным процессом. При ее протекании происходит наложение друг на друга вышеперечисленных стадий процесса. При взаимодействии исходного вещества с газом или если продуктом является газ, может возникать внешнедиффузионное или внутридиффузионное торможение. Внешнедиффузионное торможение возникает, когда газообразный исходный реагент или продукт реакции с низкой скоростью подводится или отводится от поверхности реакции. На кривой скорость реакции (d/d) – время образуется площадка. Однако при увеличении скорости газового потока внешнедиффузионное торможение снимается, и кривая 1 преобразуется в кривую 2. Внутридиффузионное торможение возникает в том случае, когда скорость диффузии в порах значительно ниже скорости диффузии газообразных веществ на поверхности вещества, газы поглощаются в устье пор. За счет этого происходит снижение скорости реакции. Чтобы снять внутридиффузионное торможение применяют размельчение частиц, либо используют частицы с более крупными порами, при этом кривая 1 преобразуется в кривую 2. Было обнаружено, что иногда продукты распада при топохимической реакции ускоряют скорость разложения. Это явление проявляется сильнее для измельченных кристаллов, чем для не измельченных. Явление ускорения реакции разложения продуктами реакции называется автокатализом. Фундаментальной характеристикой кинетики твердофазной реакции является энергия активации. Эта оценка широко используется для оценки реакций в газовой и жидкой фазах. Для анализа кинетики реакций в твердой фазе используют экспериментальные данные, пользуясь уравнением k = k0 exp (-E/RT.) Для этого находят зависимость ln k – f(1/T), E = -R [ dlnk/d(1/T)], dlnk/d(1/T) – тангенс угла наклона прямой. Для топохимических реакций энергия активации рассчитывается на некий условный моль. По значению энергии активации судят о том, в какой области протекает процесс. Если энергия активации составляет 5-20 кДж/моль, то процесс лимитируется диффузией, 20-50 кДж/моль переходная область, при энергии активации составляет 50-200 кДж/моль – процесс протекает в кинетической области. Наличие дефектов в структуре материала снижает энергию активации процесса. На скорость реакции влияет размер реагирующих частиц, при измельчении частиц увеличивается поверхность контакта, и следовательно скорость реакции. Введение в состав исходного вещества микродобавок некоторых веществ, называемых легирующими или модифицирующими, также может изменить ход реакции. По правилу Вант-Гоффа, повышение температуры на 10 градусов увеличивает скорость реакции в 2-4 раза. Еще одна возможность активирования реакционных смесей в процессе твердофазного взаимодействия заключается в изменении состава газовой среды. Если реакционные смеси содержат элементы с переменной валентностью, то при изменении окислительного потенциала газовой среды изменяется и состав твердофазных реагентов. Эффективным средством активирования твердофазных процессов является термомеханическая обработка материалов, заключающаяся в одновременном воздействии температуры и давления (прессование). Предполагают, что за счет высокой пластичности материала при повышенных температурах увеличивается площадь контактов между частицами. Например, при получении твердых растворов или ферритов смешивают оксиды или соли нескольких металлов и спекают при повышенных температурах, для улучшения взаимодействия, смесь предварительно прессуют. Процессы лимитируемые скоростью образования зародышейОбразование зародышей новых фаз является обязательной стадией очень многих реакций в твердой фазе и часто может определять их кинетику. Это относится прежде всего к реакциям разложения карбонатов, сульфатов и т. д., изучению которых большое внимание уделяется в СССР, в частности Болдыревым и Павлюченко. Эти реакции являются автокаталитическими и относятся к классу так называемых быстрых реакций в твердых телах, многие из которых, особенно если они сопровождаются выделением энергии, протекают взрывообразно. Для описания кинетики реакций разложения, контролирующийся скоростью образования зародышей, в разное время предлагались различные уравнения. Льюис, затем Центнершвер и Брюсе, Макдональд и Хиншель-вуд безуспешно пытались распространить на эти процессы уравнения кинетики гомогенных, в частности гомогенных автокаталитических, реакций. Рогинский и Шульц, считая, что 1) скорость реакции в твердых телах, идущей через возникновение и рост ядер кристаллического продукта, пропорциональна межфазовой поверхности, на которой локализуется процесс, 2) площадь этой поверхности пропорциональна произведению площадей поверхности исходного вещества и продукта, а также полагая,что S = KV2/3 (где S — площадь поверхности и V — объем фазы), получили для описания кинетики этой реакции выражение где G — доля прореагировавшего вещества. Справедливость уравнения (4) была подтверждена для процессов разложения КМn04, СаС03-6Н20, оксалата цинка и некоторых других веществ. Тем самым был подтвержден факт локализации подобного рода процессов на поверхности раздела фаз. Впоследствии Ерофеевым было показано, что уравнение (4) не является универсальным и справедливым для всех реакций, идущих через возникновение и рост ядер твердого продукта. Для описания кинетики таких реакций Ерофеев предложил уравнение которое можно приблизительно выразить также в виде Эти уравнения выведены в предположении, что процесс возникновения начальных центров реакции может быть многостадийным, т. е. что скорость этого возникновения может зависеть от времени (поскольку концентрация реагирующего вещества не остается постоянной в процессе реакции). Уравнение (5) в развернутом виде выглядит следующим образом: где v0 — число потенциальных мест, на которых могут возникать начальные центры реакции; о — число промежуточных стадий в процессе образования начальных центров; U — линейная скорость роста ядер реакции; Ki, К2. Кσ — константы, характеризующие кинетический порядок промежуточных стадий образования начальных центров реакции. Гетерогенные процессыК гетерогенным процессам относятся процессы в системах: газ – твёрдое вещество, газ – жидкость, жидкость – твёрдое вещество, твёрдое – твёрдое, жидкость – жидкость (если жидкости несмешивающиеся), газофазные процессы при участии твёрдых катализаторов. Отличительной особенностью любого гетерогенного процесса является наличие поверхности раздела фаз, которая может быть постоянной, либо меняться во времени. При протекании гетегогенного процесса наряду с чисто химическими стадиями существуют диффузионные стадии. Поэтому для управления гетерогенным процессом важна идентификация лимитирующей стадии. В общем случае для идентификации лимитирующей стадии исследуют зависимость скорости реакции от температуры, и на этой зависимости можно выделить три области: 1 – скорость возрастает с увеличением температуры, и выполняется закон Аррениуса. Это кинетическая область протекания процесса. Управляющими будут микрокинетические факторы (температура, давление, концентрации). 3 – скорость процесса практически не зависит от температуры. Диффузионная область энергии активации и диффузии очень мала, следовательно, изменение температуры не приводит к изменению коэффициента диффузии, и скорость изменяется несущественно. Управляющими являются макрокинетическими параметры, связанные со скоростью подачи реагентов, степенью перемешивания реагентов, степенью диспергирования реагентов. Также в этой области в соответствии со скоростью диффузии по первому закону Фика управляющей является концентрация реагентов: 2 – переходная область. Скорость увеличивается с увеличением температуры, но закон Аррениуса не выполняется. В этой области управляющими являются и микро- , и макрокинетические факторы, но интенсивность их воздействия на процесс меньше, чем в соответствующей области протекания. С точки зрения промышленной реализации область 2 наименее перспективна, но следует учитывать, что по ходу гетерогенного процесса он может переходить из одной области в другую. Поэтому для предотвращения перехода изменение одного из микрокинетических параметров обычно сопровождают изменением какого-либо макрокинетического параметра. Гетерогенный процесс – это процесс многостадийный. Наиболее простыми являются процессы в системе жидкость – газ, которые обычно протекают в три стадии: 1-я стадия: область внешней диффузии, то есть подвод газа и жидкости к поверхности раздела фаз, которая чаще всего формируется искусственно (насадочная колонна). 2-я стадия: химическое взаимодействие. Кинетическая область. 3-я стадия: отвод продуктов от поверхности раздела фаз. Внешняя диффузионная область протекания процесса. В подавляющем большинстве случаев процессы в системе жидкость – газ протекают во внешней диффузионной область, поэтому при проектировании оборудования необходимо решать проблему одновременного увеличения линейной скорости подачи реагентов и увеличение площади поверхности насадки. Для того чтобы создать требуемую поверхность контакта фаз необходимо уменьшать размер элементов насадки, что приводит к увеличению гидравлического сопротивления при увеличении скорости подачи реагентов. Следовательно, необходимо искать оптимум в этом вопросе. При проведении адсорбции температура не является управляющим параметром, так как при увеличении температуры растворимость газа в жидкости уменьшается, и увеличение температуры начинают только в том случае, если общая скорость абсорбции лимитируется химической реакцией. Наибольшую сложность для рассмотрения представляют процессы в системе газ – твёрдое вещество. В общем случае процесс можно представить как совокупность 11 стадий: 1-я стадия: диффузия газообразного реагента к поверхности твёрдой частицы (внешняя диффузия). 2-я стадия: диффузия газообразного реагента через слой продукта к поверхности раздела фаз (внутренняя диффузия). 3-я стадия: адсорбция газообразного реагента на поверхности раздела фаз. 4-я стадия: растворение газообразного реагента в твёрдом непрореагировавшем исходном веществе. 5-я стадия: диффузия от поверхности раздела фаз к потенциальному центру образования ядра новой фазы. 6-я стадия: химическая реакция. Далее в обратной последовательности (5-я, 4-я, 3-я, 2-я, 1-я расшифровать). Все 11 стадий наблюдаются в том случае, если уравнение реакции имеет вид: Атв + Вгаз = Ств + Dгаз. Если же: Атв = Ств + Dгаз , то шесть стадий, начиная от химической реакции (с 6-ой по 11-ю). Если: Атв + Вгаз = Ств , то шесть стадий, начиная с диффузии газообразного реагента к поверхности твёрдой частицы (с 1-ой по 6-ю). Для описания кинетики твёрдофазного взаимодействия используется три основные модели образования ядер новой фазы. Образование ядер новой фазы происходит с одинаковой вероятностью на всей внешней поверхности твёрдой частицы при реализации физических условий процесса. Такая модель может быть применена при рассмотрении процессов разложения твёрдого материала, если температура процесса выше, чем температура начала разложения. В этом случае при реализации физических условий процесса вся поверхность твёрдой частицы покрывается слоем продукта, и дальнейшее продвижение к поверхности раздела фаз обуславливается только диффузионными сопротивлениями, обусловленными как пористостью материала, так и размером твёрдых частиц. Образование ядер новой фазы на активных центрах происходит с одинаковой вероятностью. В качестве активных центров рассматривают дефекты кристаллической решётки твёрдого материала и включения микропримесей, которые обязательно присутствуют в материале. Согласно этой модели считается, что активные центры равномерно распределены по поверхности твёрдой частицы. И при реализации физических условий процесса на поверхности твёрдой частицы образуется фиксированное количество ядер новой фазы. Далее наблюдается рост ядер, что в начальный период времени приводит к увеличению поверхности раздела фаз, а в дальнейшем к её уменьшению. Математически эта модель описывается уравнением сжимающейся сферы: где: x – степень превращения твёрдого материала, k – константа скорости в соответствии с уравнением Аррениуса, Модель сжимающейся сферы наиболее хорошо описывает процессы разложения твёрдого материала и некоторые процессы, связанные с присоединением газообразного реагента. Модель экспоненциального роста числа ядер новой фазы. Универсальна, описывает любой процесс. Предполагает, что активные центры на поверхности твёрдой частицы энергетически неоднородны. При реализации физических условий процесса ядра новой фазы образуются на активных центрах, обладающих наибольшей избыточной энергией. Появление поверхности раздела фаз приводит к активации центров, обладающих меньшей избыточной энергией в первоначальный момент времени. Зависимость скорости процесса от времени обработки твёрдого материала. Участок О–А: индукционный период. Он предназначен для накопления энергии в твёрдом веществе. Считается, что в течение индукционного периода протекают первичные превращения, приводящие к возникновению первых ядер новой фазы. Очевидно, что продолжительность индукционного периода зависит от температуры. Участок А–С: период ускорения. На этом участке протекают два параллельных процесса: образование ядер новой фазы и рост уже образовавшихся. Разбит на две части, чтобы показать, что на начальном этапе (участок А–В ) рост связан именно с увеличением количества ядер новой фазы, а на участке В–С – с ростом ядер. Участок С–Д : период максимальной скорости. В точке максимума прекращается образование ядер новой фазы. Участок Д–Е: период спада. В точке Д растущие ядра начинают соприкасаться между собой: из плоских превращаются в шарообразные. С точки Е процесс переходит в диффузионную область. С этой точки вся поверхность частицы покрыта ядрами новой фазы. Эта модель описывается уравнением Ерофеева: n – постоянная Ерофеева. Её физический смысл связан с количеством ядер новой фазы, которое активирует одно образовавшееся ядро. Эта величина определяется экспериментально. Кинетические модели топохимических реакций.Топохимические реакции – химические реакции, происходящие в твердой фазе, при которых процесс локализован на границе раздела твердое исходное вещество – твердый продукт реакции. Примеры топохимических реакций: дегидратация кристаллогидратов, окисление металлов, реакции процессов обжига, разложение взрывчатых веществ и др. название «топохимические» было предложено в 1918 г. Кольшуттером и в буквальном переводе означает: реакции, протекание которых связано с определенным местом в кристалле (от греческого «topoz» – место). Основные черты топохимических реакций в значительной мере определяются тем, что реагирующие атомы, молекулы или ионы, образующие кристалл, жестко закреплены в узлах кристаллической решетки и лишены той подвижности, которой они обладают в газовой или жидкой фазах. Реакционная способность атомов или ионов в сильной степени зависит от того, в каком месте кристалла они находятся (в объеме, на поверхности грани, на ребре кристалла, на его вершине или на пороге дислокации). В связи с этим, при топохимических реакциях существенное значение приобретает реальная структура кристалла, связанная с наличием в кристалле различного рода дефектов, а образующийся при реакции твердый продукт, находясь в контакте с непрореагировавшим исходным веществом, может оказывать на скорость реакции каталитическое влияние, сила которого зависит от состояния продукта реакции. Благодаря тому, что при топохимических реакциях допустимо лишь минимальное перемещение реагирующих частиц, очень часто продукт реакции сохраняет внешнюю форму кристаллов исходного вещества. Так как процесс локализован в пределах реакционной зоны, скорость процесса пропорциональна величине реакционной зоны в данным момент времени. Следовательно процесс лимитируется образованием центров кристаллизации (твердого продукта реакции). Первоначально, как только центры реакции образовались, скорость реакции невелика, но по мере их роста, скорость – возрастает. Начиная с какого-то момента времени, зародыши начинают пересекаться. Момент их пересечения соответствует максимальной скорости. После пересечения площадь реакционной зоны начинает сокращаться, при этом уменьшается скорость.
Рисунок 31. а – зависимость доли прореагировавшего вещества от времени; б – зависимость скорости реакции от времени. На основании таких простых представлений о развитии топохимических реакций было выведено большое количество кинетических уравнений, наиболее простым из них является уравнение, предложенное Рогинским и Шульцем. Если число зародышей после того, как началась реакция, остается постоянным, скорость реакции должна быть пропорциональна реакционной поверхности S каждого зародыша. Так как поверхность двумерна S = [l] 2 , а объем (и следовательно масса, которая пропорциональна объему) трехмерен v = [l] 3 , то выражая l через v и, соответственно, через степень прореагировавшего вещества a, получим: Согласно этому уравнению, скорость реакции пропорциональна доли прореагировавшего вещества в степени 2/3, а общее количество прореагировавшего вещества пропорционально кубу времени. Уравнение (126) было применено для анализа кинетических данных по термическому разложению твердых веществ. Процесс разложения азида кальция описывается на 40%, а a-азида свинца на 50%. Из самого смысла уравнения (126) следует, что оно может быть применено лишь в первой стадии реакции – до максимума скорости. После пересечения зародышей реакционная поверхность будет меньше, чем это следует из суммирования реакционных поверхностей отдельных зародышей, и поэтому начиная с этого момента, уравнение (126) будет давать завышенные значения скорости реакций. Слияние реакционных поверхностей отдельных зародышей приводит к образованию общей реакционной поверхности, которая пропорциональна поверхности непрореагировавшей части кристалла. Выражая эту поверхность через массу непрореагировавшего вещества, получим следующее уравнение где а – масса непрореагировавшего вещества к моменту времени t. Если, к примеру, в результате слияния зародышей образуется реакционная зона сферической формы, то скорость процесса хорошо описывается уравнением сокращающейся сферы Эти простые уравнения применяют при описании кинетики разложения многих веществ, однако экспериментально многие процессы не описываются этими уравнениями. Существует целый класс реакций, для которых число зародышей N определяется уравнением где n – показатель степени, для быстрых экзотермических реакций n = 4-8 до 20. Он зависит от геометрической формы зародышей (куб, плоскость, сфера и т.д.). Кинетика подобных реакций хорошо описывается обобщенным уравнением кинетики топохимических реакций выведенное Ерофеевым в 1946г. где p – вероятность реагирования. Позднее это уравнение приведено к наиболее известному в настоящее время виду топокинетического уравнения применительно к реакциям твердых веществ Это уравнение известно как Уравнение Казеева – Ерофеева – Колмогорова. По Ерофееву n – число последовательных стадий при образовании устойчивого начального центра новой фазы, плюс постоянное число, характеризующее форму зародыша, и равное 3 при образовании сферического зародыша, 2 – цилиндрического и 1 – плоского. Из смысла уравнения следует, что сопоставление скоростей по константе скорости может быть сделано лишь в том случае, если n постоянно, что наблюдается далеко не всегда. По смыслу константа скорости должна иметь ту же размерность, что и константа скорости мономолекулярной реакции. В уравнении (130) постоянная k зависит от n. В общем случае величина n отражает характер развития реакционной зоны. Во всех случаях. Когда n>1, исследуемый процесс находится в кинетической области. При n Дата добавления: 2014-12-26 ; просмотров: 4230 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ Изучение топохимической реакцииАвтор работы: Пользователь скрыл имя, 23 Марта 2012 в 14:17, курсовая работа ОписаниеКатализ (от греч. katálysis — разрушение), изменение скорости химических реакций в присутствии веществ, вступающих в промежуточное химическое взаимодействие с реагирующими веществами, но восстанавливающих после каждого цикла промежуточных взаимодействий свой химический состав. Реакции с участием катализаторов называются каталитическими. Количество реагирующего вещества, которое может испытать превращение в присутствии определённого количества катализатора, не ограничивается какими-либо стехиометрическими соотношениями и может быть очень большим. Работа состоит из 1 файлвся лаба.docxЕще в древние времена было известно ноющее действие золы растений, которое- связано с присутствием в ней карбоната калия. Получение карбоната калия из древесной золы было известно уже древним грекам и римлянам, однако они не отличали полученную таким образом соль от встречающейся в природе соды. Алхимики готовили чистый карбонат калия прокаливанием виннокаменной соли , а позднее путем сжигания ее с селитрой. Для получения поташа Леблан разработал способ, совершенно аналогичный его способу получения соды. Долгое время этот способ имел большое техническое значение. Карбонизация калийного щелока происходит непосредственно вслед, за его электролитическим получением при пропускании через него двуокиси углерода По предложению Харгревса (Hargreaves) двуокись углерода вводят непосредственно в камеру для электролиза, так что там тотчас образуется К2С03; однако в этом случае поташ трудно отделить от хлорида. Стассфуртский способ [способ Энгела (Engel) и Прехта (Precht)] основан на плохой растворимости двойной соли гидрокарбоната калия и карбоната магния. По этому способу двуокись углерода пропускают в раствор хлористого калия, в котором суспендирован: Выпадающая труднорастворимая двойная соль разлагается в воде при температуре около 60° с выделением двуокиси углерода По формиатному способу [способ Гольдшмидта (Goldschmidt)] поташ получают через формиат калия , для чего раствор сульфата смешивают в молярном отношении с известковым молоком, и эту смесь насыщают окисью углерода при повышенной температуре, (около 220°) и давлении 30 am. Реакция идет по уравнению в результате чего образуется раствор формиата калия, который после отделения гипса упаривают досуха. Методом окислительного кальцинирования получают затем: карбонат по уравнению Карбонат калия применяют в мыловаренном и стекольном производствах, при крашении, белении и отмывке шерсти. Его используют также для получения цианида калия, а в препаративной химии — часто в качестве водуотнимающего средства. Гидрокарбонат калия. Кислый или первичный карбонат калия несколько более трудно растворим, чем нейтральный карбонат калия, но существенно легче растворим, чем гидрокарбонат натрия (а именно 36,1 г в 100 з воды при 26°). Он имеет бесцветные моноклинные кристаллы удельного веса 2,17. При нагревании он распадается так же, как гидрокарбонат натрия: Карбонаты рубидия и цезия. и удобнее всего получать взаимодействием сульфатов с гидроокисью бария и последующим упариванием с карбонатом аммония. Обезвоженные прокаливанием соли расплываются на воздухе и растворяются в воде со значительным выделением тепла (8,75, соответственно 11,84 ккал). В спирте карбонат рубидия, так же как другие карбонаты щелочных металлов, незначительно растворим, карбонат цезия, напротив, легко растворим. В 100 г спирта при 19° растворяется 0,74 г и 11,1 г . Это используют для разделения рубидия и цезия. Гидрокарбонаты рубидия и цезия еще лучше растворимы, чем гидрокарбонат калия. Карбонат лития. выделяется при смешивании растворов солей лития с растворами, содержащими ионы в виде труднорастворимой в воде и нерастворимой в спирте белой кристаллической соли. Удельный вес его 2,11. Точка плавления 618°. При техническом получении карбоната лития большей частью исходят из амблигонита , встречающегося в виде больших залежей в Америке. Его растворяют в горячей концентрированной серной кислоте. Примеси алюминия и железа осаждают добавлением аммиака и сульфида аммония и водную вытяжку смешивают с карбонатом натрия. Выделившийся при этом карбонат лития очищают растворением в соляной кислоте, осаждением примесей серной кислоты хлоридом бария и повторным осаждением соли в виде карбоната. Получение лития из других минералов проводят аналогично, с той только разницей, что вскрытие производят по-другому — например, в случае сподумена путем сплавления с содой. Силикаты можно вскрывать также прокаливанием с или с . Для вскрытия трифилина лучше всего применять концентрированную серную кислоту с небольшим добавлением азотной кислоты. Отделение лития от других щелочных металлов можно производить, используя плохую растворимость фосфата в воде или определенную растворимость хлорида в смеси спирта с эфиром. Карбонат лития лучше растворим в холодной воде, чем в горячей. При нагревании от 0 до 100° растворимость падает от 1,54 до 0,73 ч. на 100 ч. воды. Растворимость значительно увеличивается при добавлении в воду . Этим иногда пользуются для очистки карбоната. Можно предположить, что в воде, содержащей , образуется гидрокарбонат, так же как в случае щелочноземельных металлов. В твердом состоянии гидрокарбонат лития не получен. Карбонат лития является исходным продуктом для получения большинства других солей лития. Раисе его применяли, для лечения подагры. Теперь для этой цели используют органические соли лития, такие, как цитрат лития или салицилат лития. Образование и диссоциация карбонатов, восстановление окислов и сульфидов, производство цемента, огнеупоров, керамики и многие другие химические превращения являются сложными гетерогенными процессами, имеющими огромное практическое значение. Их особенность состоит в том, что химическая стадия процесса сопровождается превращением в твердом состоянии, когда вещество с одной кристаллической решеткой исчезает и образуется продукт с другой структурой. Простейшим примером такой реакции является диссоциация Атв →Втв + Сгаз. Указанные реакции называют топохимическими. Для них характерно нарастание скорости в начальный период процесса и достижение ею предельного значения с последующим постепенным спадом до нуля (рисунок 1). Одним из внешних признаков топохимической реакции служит S –образный вид кинетической кривой (рисунок 1, кривая 1). Рисунок 1 — Кинетические зависимости топохимической реакции Топохимическая реакция начинается обычно на отдельных участках поверхности, в так называемых потенциальных центрах реакции, откуда она постепенно распространяется в глубь кристалла. Центрами реакции являются места на поверхности кристалла, где связи частиц в решетке ослаблены (вершины углов, ребра, дефекты на поверхности). В первый период реакции образуются микроскопические зародыши ядер новой фазы (реакция идет с ничтожной скоростью). Образование зародышей ядер, в свою очередь, вызывает искажение материнской решетки, которое способствует возникновению новых зародышей. Одновременно увеличиваются и сами ядра, поверхность реакции растет, в результате растет скорость химического взаимодействия. Этот период является автокаталитическим, так как образование продукта реакции, приводящее к увеличению поверхности раздела фаз, вызывает возрастание скорости. Скорость процесса будет максимальной при наибольшей величине поверхности реакции, когда межфазные границы отдельных зон, центров приходят в соприкосновение. После того, как зоны перекрываются, поверхность начинает уменьшаться и скорость реакции падает. Для того чтобы получить уравнение кинетики топохимической реакции, нужно знать или предположить, по крайней мере, следующие основные параметры: — закон образования ядер новой фазы; — закон роста ядер; Возможны различные сочетания законов образования, роста и формы ядер продукта, поэтому существует большое количество уравнений, описывающих кинетику топохимических реакций. Но наиболее широко применяются лишь несколько из них. На основании вероятности взаимодействия молекул данной системы и вне связи с предложениями об истинном механизме реакции Ерофеев получил уравнение где α — доля прореагировавших молекул к моменту времени t; р – вероятность того, что молекулы прореагируют в течение времени от t до t + dt. Определив р, можно получить кинетическое уравнение соответствующего процесса. В общем случае вероятность взаимодействия молекул в любой момент времени пропорциональна суммарной поверхности реакции, т.е. Р = А. Примем, что поверхность реакции изменяется во времени по степенному закону где A, A′, A″ — коэффициенты пропорциональности. Здесь к и m – эмпирические коэффициенты, не имеющие четкой физической интерпретации. Значению к иногда приписывается смысл константы скорости. Величины к и m находят графически по экспериментальным данным. Для этого уравнение представляют в линейной форме Lnln[1/(1-a)] = lnk + mlnt. И по графику в координатах lnln[1/( 1-a)], lnt определяют к и m. 2.2 Методики эксперимента В лаборатории столы старые, но удобные для проведения опытов. Для каждой лабораторной работы приготовлены реактивы, написан номер работы, если студент пропустил выполнение работы, он может в дни консультаций её отработать под руководством лаборанта. На столе штатив с пробирками, держатели для пробирок. Реактивы на полках. В лаборатории за каждым столом работает по 3-4 студента, делают опыты и в тетрадях записывают наблюдения и выводы. Остальная часть работы оформляется в кабинете. Это наша установка! «Тут мы работаем с различными химическими реактивами, здесь очень интересно проводить опыты и наблюдать за реакциями. Так же здесь большое количество банок для опытов. Цветы, которые стоят полках, скрашивают лабораторию, её серые тона. На столах стоят пробирки. Лаборатория — это то место, где интересно и познавательно!» Стенды и оформление лаборатории Основные требования к промышленным катализаторам Катализаторы являются «сердцем» каталитических процессов, и они должны удовлетворять многим требованиям, которые относятся к их эксплуатационным свойствам, активности и селективности, текстуре и другим свойствам. При разработке новых катализаторов необходимо создавать наиболее простые из них, необходимо учитывать тип химического процесса и тип промышленной установки, ее конструкцию и производительность. Катализатор занимает малую долю в затратах на переработку сырья, однако экономическую сторону при создании нового катализатора и методов его производства необходимо также учитывать. При создании нового твердого катализатора или усовершенствовании находящегося в эксплуатации катализатора необходимо учитывать следуюшие основные параметры для катализаторов:
К физико-механическим свойствам или параметрам катализатора можно отнести насыпную плотность, истинную плотность, удельную поверхность, средний объем пор и распределение пор по радиусам, фракционный состав, размер частиц, пористость, аморфность или кристалличность, форму частиц, теплоемкость, термостойкость или водо-паротермостойкость, способность к отравлению и регенерации. К химическим параметрам катализаторов можно отнести химический состав, содержание примесей, способность к активированию (промотированию, модифицированию) и отравлению ядами, образованию сплавов, модификаций и фаз, прививке активаторов к поверхности твердых катализаторов. Эксплуатационно-экономическими показателями или свойствами катализаторов являются активность и селективность, легкая регенерируемость от различных отложений и включений (кокса, оксидов, обратимых ядов), возможность создания простых способов синтеза катализатора в промышленном масштабе, повышенная теплоемкость, насыпная плотность, малая чувствительность к ядам, длительное время работы в реакторе без регенерации, легкость перевозок и хранения, легкость отделения от реакционной смеси, доступность сырья для производства катализатора и экологическая безвредность. Важную роль в подборе, синтезе и эксплуатации катализаторов играют теории катализа и катализаторов, которые позволяют оптимизировать многие параметры для катализаторов, их производства и эксплуатации. Физические свойства адсорбентов и катализаторов Физические свойства адсорбентов и катализаторов объединяют такие параметры как: пористость, фракционный состав, насыпная, кажущаяся и истинная плотности, механическая прочность, удельная поверхность, распределение пор по радиусам, объем пор. теплоемкость, теплопаростойкость. Пористость адсорбентов и катализаторов Твердые адсорбенты и катализаторы готовят, как правило, в форме цилиндриков, колец Рашига, шариков, микросфер, частиц в форме звездочек, дужек, лепешек и многих других форм. Металлические катализаторы готовят в форме сеток или свитых проволочек. Как правило твердые оксидные, сульфидные и другие частицы катализаторов пронизаны порами. Поры могут иметь разную форму. Они могут полностью проходить сквозь частицу катализатора или иметь тупиковую форму, могут быть прямыми, изогнутыми или зигзагообразной формы. Катализаторы могут быть тонкопористыми, широкопористыми и со смешанным набором пор. |





 . Отличительный признак диффузионной области: первый порядок реакции по компоненту независимо от молекулярности реакции.
. Отличительный признак диффузионной области: первый порядок реакции по компоненту независимо от молекулярности реакции. ,
, — время обработки твёрдого материала.
— время обработки твёрдого материала.
 ,
,

 a da/dt
a da/dt
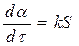 (125)
(125)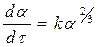 (126)
(126)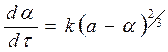 , (127)
, (127)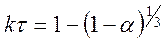 .
.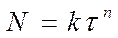 (128)
(128)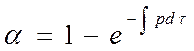 (129)
(129)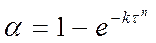 (130)
(130)