ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Конспект лекций для студентов биофака ЮФУ (РГУ)
2.1 СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
2.1.9 Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Я. Г. Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ . Математически правило Вант-Гоффа можно записать следующим образом:
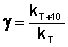 (II.29)
(II.29)
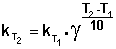 (II.30)
(II.30)
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
2.1.10 Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. С. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом:
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е’А выше, нежели энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
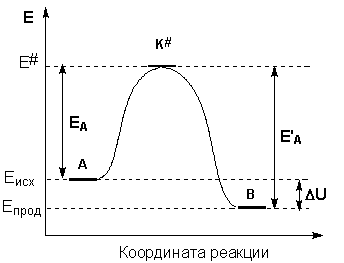
Рис. 2.5 Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.6):
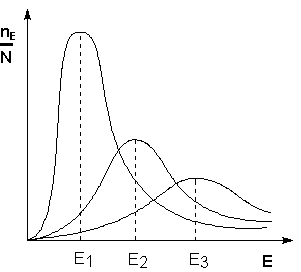
Рис. 2.6 Распределение частиц по энергии
Здесь nЕ/N – доля частиц, обладающих энергией E;
Ei — средняя энергия частиц при температуре Ti (T1 уравнения Аррениуса . Согласно уравнению изобары Вант-Гоффа,
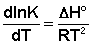 (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
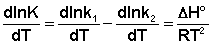 (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
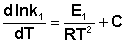 (II.33)
(II.33)
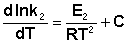 (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации :
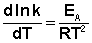 (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
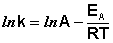 (II.36)
(II.36)
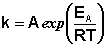 (II.37)
(II.37)
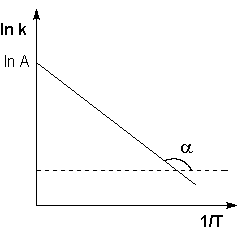
Рис. 2.7 Зависимость логарифма константы скорости химической
реакции от обратной температуры.
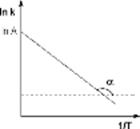
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
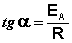 (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
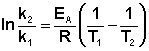 (II.39)
(II.39)
Copyright © С. И. Левченков, 1996 — 2005.
- График Аррениуса и определение энергии активации температурных реакций
- Химия.. Теория переходного состояния (активированного комплекса)
- 19. Теория активированного комплекса. Энергия активации. Уравнение Аррениуса. Энергетическая диаграмма реакции.
- Суть теории переходного состояния (активированного комплекса):
- Энергия активации
График Аррениуса и определение энергии активации температурных реакций
Уравнение Аррениуса – это температурная зависимость константы скорости К элементарной химической реакции:
где A – предэкспоненциальный множитель (размерность совпадает с размерностью К), Еа-энергия активации, обычно принимающая положительные значения, Т – абсолютная температура, k-постоянная Больцмана. Принято приводить Еав расчете не на одну молекулу, а на число частиц NA= 6,02*1023(постоянная Авогадро) и выражать в кДж/моль; в этих случаях в уравнении Аррениуса величину k заменяют газовой постоянной R.
График зависимости 1nК от 1/kT (график Аррениуса) – прямая линия, отрицательный наклон которой определяется энергией активации Еа и характеризует положительную температурную зависимость К.
Энергия активации в элементарных реакциях, минимальная энергия реагентов (атомов, молекул и др. частиц), достаточная для того, чтобы они вступили в химическую реакцию, т. е. для преодоления барьера потенциальной энергии на поверхности, отделяющего реагенты от продуктов реакции. Потенциальный барьер — максимум потенциальной энергии, через который должна пройти система в ходе элементарного акта химического превращения. Высота потенциального барьера для любого пути, проходящего через переходное состояние, равна потенциальной энергии в переходном состоянии. Если в сложной реакции, состоящей из последовательных и параллельных элементарных реакций, имеется лимитирующая элементарная реакция (реакция с максимальным характерным временем), то ее энергия активации является и энергией активации сложной реакции. В макроскопической химической кинетике энергия активации – энергетический параметр Еа, входящий в уравнение Аррениуса К=Аexp(-Еа / RТ). Наблюдаемая энергия активации вычисляется из тангенса угла наклона прямой графика Аррениуса. В общем случае сложных реакций параметр Еа в уравнении Аррениуса является функцией энергии активации отдельных стадий, и определяемая энергия активации называется эффективной (эмпирической, кажущейся). Любой процесс, сопровождающийся каким-либо изменением энергии, является экзотермическим в одном направлении и эндотермическим в другом. Энергии активации экзотермического и эндотермического направлений реакции, обозначаемые соответственно Е — а и Е + а , связаны соотношением: Е + а=Е — а+|Q|, где Q — .теплота реакции при Т=0. Качественная одномерная геометрическая иллюстрация связи энергии активации с высотой потенциального барьера и теплотой реакции представлена на рисунке, где q -координата реакции; Е1 и Е2 — уровни энергии соответственно основного состояния реагентов и продуктов реакции.
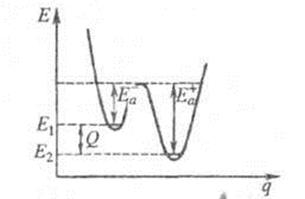
Энергетическая схема элементарной реакции.
Аналитическое применение методов ДТА и ДСК.
Позволяет проводить анализ изменения массы и тепловых потоков в одном эксперименте для одного и того же образца. Сочетая методы ТГА (термографический анализ) и ДСК, можно определить следующие характеристики материалов: с помощью ТГА фиксируется изменение массы образца при программируемом изменении температуры, при этом так же определяется массовый остаток вещества после термовоздействия.
С помощью ДСК определяются следующие характеристики: теплота плавления кристаллических веществ, удельная теплоемкость в широком диапазоне температур, температура стеклования полимерных материалов, степень кристалличности энтальпия реакции термостойкость и устойчивость к окислению старение материалов чистота веществ фазовые переходы, полиморфные превращения идентификация продукта по набору теплофизических характеристик.
Можно проводить анализ:
· температуры фазовых переходов
· поведения при кристаллизации
Лекция 6.
Термогравиметрия.
Наряду с методом дифференциально-термического анализа веществ активно развивалась и вторая ветвь термического анализа – метод термогравиметрии. С помощью последнего можно с высокой степенью точности проследить за изменением массы пробы при повышении температуры.
Термогравиметрия – это метод исследования, заключающийся в измерении изменения массы образцов при нагревании. Первоначальную схему метода можно представить следующим образом: пробу нагревали до определенной температуры, затем охлаждали и после охлаждения взвешивали с аналитической точностью. Процесс повторяли циклически, каждый раз увеличивая температуру. Если результаты взвешивания, относящиеся к отдельным температурным значениям, представить в координатах температура – масса образца и соединить полученные точки, то получится кривая, именуемая термогравиметрической (ТГ). Описанный метод является исключительно длительным и неточным, но применяется и сегодня, например, при аналитическом определении потери массы при прокаливании вещества. Значительно быстрее и точнее проводить измерения с помощью термовесов, непрерывно регистрирующих изменение массы пробы.
Принцип работы термовесов следующий. Пробу помещают в тигель (рис. 3), опирающийся на коромысло весов. Затем тигель нагревают в электрической печи так, чтобы его температура равномерно повышалась. Температура печи измеряется с помощью находящейся в ней термопары, к концам которой подключен милливольтметр, и время от времени (например, каждые 5. 10 К) масса образца фиксируется.
Графически изображенные результаты измерения дают термогравиметрическую кривую (рис. 4). Если изменение массы регистрируется автоматически, кривая ТГ строится в зависимости не от температуры, а от времени, однако такая замена оси абсцисс обратима, если одновременно фиксируется и зависимость температуры в печи от времени. Наиболее просто замена оси абсцисс осуществляется в том случае, когда повышение температуры в печи происходит равномерно во времени.
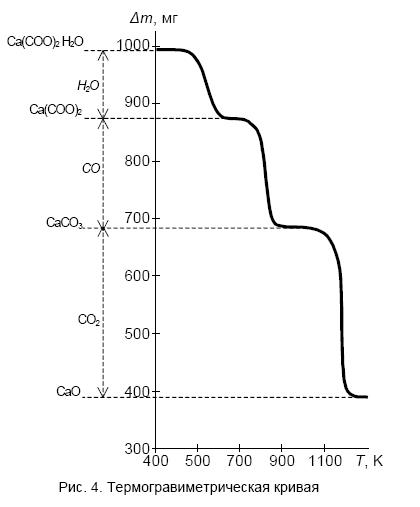
На основании кривой ТГ можно судить о том, каким образом изменялась при нагревании масса пробы, например, при каких температурах и на сколько миллиграммов менялась масса пробы осадка ацетата кальция, а следовательно, при каких температурах происходили химические превращения Ca(СОО)2*H2O → Ca(СОО)2 → CaСО3 → CaО.
Степень изменения массы определяется в зависимости от типа термовесов с точностью, примерно, от 0.1 до 0.5%, поэтому на основании результатов измерения можно производить довольно точные стехиометрические расчеты (стехиометрия – наука о соотношении массовых или объемных долей реагирующих веществ).
Конструкция термовесов постоянно модифицировалась. Первый экземпляр термовесов сконструировал японский исследователь Гонда в 1915 г. Впоследствии многие исследователи шли по пути совершенствования именно его конструкции. Среди используемых в настоящее время есть весы, качающиеся по призмам, весы с подвешенным коромыслом, весы с тормозящей нитью, весы пружинного типа, весы, снабженные жидкостным, воздушным или электромагнитным затуханием. Но, рассуждая объективно, ни одному из многочисленных типов нельзя отдать безусловное предпочтение.
Как изучение процессов, происходящих при нагревании глинистых минералов и пород, потребовало широкого распространения и развития метода ДТА, так и нерешенные вопросы определения постоянного состава аналитических осадков ускорили распространение метода термогравиметрии. Для исследования же иных вопросов последний метод долгое время применялся очень редко.
Недостатки термогравиметрии обнаруживаются только тогда, когда целью испытания является определение хода процесса разложения. Кроме того, в тех случаях, когда две реакции следуют плотно друг за другом либо перекрывают друг друга или же чередуются реакции с большими и небольшими изменениями массы, тогда метод термогравиметрии оказывается неопределенным и оценка кривой становится затруднительной и неточной.
Указанные трудности попытались устранить конструированием вакуумных термовесов. Сущность термогравиметрических испытаний в вакууме заключается в том, что выделившиеся газообразные побочные продукты немедленно удаляются из внутренней части материала, вследствие чего равновесие реакций разложения смещается в сторону разложения. Между твердой и газообразной фазами всегда устанавливается равновесие, изменяющееся в соответствии с парциальным давлением газовых продуктов. Термическое разложение в вакууме обычно происходит в узких температурных пределах и быстро, поэтому плотно следующие друг за другом реакции лучше отделяются друг от друга.
Эти же проблемы вынудили исследователей разработать новый статический метод термогравиметрии, который был назван ими методом ступенчатого изотермического нагревания. Такой метод испытания несмотря на применение в нем автоматически работающих современных термовесов в действительности означал возвращение к старому методу периодического нагревания и взвешивания. Температуру печи при испытании не увеличивали до тех пор, пока масса пробы не становилась постоянной. Затем, незначительно увеличив температуру, опять дожидались постоянства массы. Таким способом удалось достичь того, что даже в случае медленно происходящих процессов устанавливалось равновесие, соответствующее данной температуре, и реакции, происходящие при более низкой температуре, не смешивались с реакциями, протекающими при более высокой. Полученные кривые показывают резкие и определенные переломы, значительно облегчающие оценку. Однако применение этого метода целесообразно лишь в исключительных случаях, так как процесс измерения является весьма длительным.
Таким образом, несмотря на всевозможные ухищрения, предпринимаемые для устранения трудностей оценки кривой ТГ, исследователям стало ясно, что необходим качественно новый подход к измерению.
Химия.. Теория переходного состояния (активированного комплекса)
| Название | Теория переходного состояния (активированного комплекса) |
| Анкор | Химия..docx |
| Дата | 21.08.2018 |
| Размер | 91.17 Kb. |
| Формат файла |  |
| Имя файла | Химия..docx |
| Тип | Документы #23315 |
Подборка по базе: Экономическая теория 2.2.docx, Мудл Функц.пробы Теория.docx, презентация Теория Проиндустриальноеобщество.pptx, Общая теория криминалистики.docx, Практическое задание Теория государства и права №4 Евдокимова И., реферат экономическая теория.docx, Задание по диаграмме состояния двойных сплавов.pdf, Эссе на тему Какой экономический рост нужен россии с учетом особ, Лекция 1 Теория СИ (2) (1).pptx, Реферат.Экономическая теория часть 1.docx19. Теория активированного комплекса. Энергия активации. Уравнение Аррениуса. Энергетическая диаграмма реакции.Теория переходного состояния (активированного комплекса) В попытках устранить недостатки теории активных столкновений ученые предложили новую теорию химической кинетики. Это сделали практически одновременно в 1935 году, более чем через полвека после открытий Аррениуса, Г.Эйринг (США) с одной стороны, а также М.Поляни и М.Г.Эванс (Великобритания) — с другой. Они предположили, что химическая реакция между началом и завершением претерпевает некое «переходное состояние», как его назвали Эванс и Поляни, при котором образуется неустойчивый «активированный комплекс» (термин Эйринга). Энергия активации как раз и требуется для достижения этого состояния, при котором вероятность успешного завершения реакции весьма велика. Поэтому энергия активации и может быть меньшей, чем энергия разрыва исходных химических связей. Суть теории переходного состояния (активированного комплекса):1)частицы реагентов при взаимодействии теряют свою кинетическую энергию, которая превращается в потенциальную, и для того чтобы реакция свершилась, необходимо преодолеть некий барьер потенциальной энергии;
|

