Количественная зависимость константы скорости реакции от температуры была впервые предложена тоже Я.Вант-Гоффом (1887) в виде уравнений изохоры и изобары химической реакции (см. п. 4.5.2).
Эта идея была развита С.Аррениусом (1889), который открыл, что температурную зависимость скорости многих реакций можно описать уравнением:
k = Ае — Е* / RT
где k — константа скорости, e — основание натуральных логарифмов, R — универсальная газовая постоянная, T – температура, А — предэкспоненциальный множитель, Е* — энергия активации реакции.
Чтобы выяснить физический смысл величин А и Е*, входящих в уравнение Аррениуса, следует сначала познакомиться с основными положениями теории активных соударений(столкновений)(С.Аррениус и Я.Вант-Гофф; 1880-е г.г.):
1) Химическое взаимодействие между молекулами возможно только при их столкновении.
2) Не каждое столкновение молекул приводит к химическому взаимодействию, т. е. является результативным или, по терминологии Аррениуса, активным. Существует некий энергетический барьер, преодолеть который и вступить во взаимодействие может лишь часть молекул, причём, как правило, это очень малая часть от их общего числа в системе.
3) Причиной, обусловливающей существование энергетического барьера, является взаимное отталкивание электронных оболочек молекул при их сближении.
Когда две частицы удалены друг от друга на очень большое расстояние, между ними нет никакого взаимодействия и потенциальная энергия такой системы равна нулю. При меньших расстояниях между частицами они притягиваются друг к другу, и потенциальная энергия системы понижается. При дальнейшем уменьшении расстояния становятся заметными силы отталкивания электронных оболочек молекул и потенциальная энергия резко возрастает. Поэтому для сближения частиц до расстояния, на котором начнется перераспределение электронов на их орбиталях (т. е. химическое взаимодействие), частицы должны обладать достаточно большим запасом кинетической энергии. Силы отталкивания между частицами и представляют между собой так называемый потенциальный или энергетический барьер, а химическое взаимодействие возможно только в том случае, если сталкивающиеся молекулы способны преодолеть его.
4) Для того, чтобы молекулы могли при столкновении преодолеть энергетический барьер, они должны двигаться навстречу друг другу с достаточно большой скоростью. Для достижения этой необходимой скорости нужна определённая энергия, называемая энергией активации. Энергия активацииЕ* — это избыток энергии активных молекул по сравнению с неактивными, или иначе, энергия, которой должны обладать молекулы, чтобы иметь возможность вступить во взаимодействие. Размерность СИ энергии активации — Дж/моль.
5) Чем больше энергия активации реакции, тем больше энергетический барьер, и тем меньшее число молекул способно его преодолеть. Поэтому, чем больше Е*, тем медленнее идёт реакция.
6) С повышением температуры увеличивается скорость теплового движения молекул, поэтому доля активных молекул возрастает. Иными словами, при повышении температуры происходит термическая активация, приводящая к увеличению скорости реакции.
Возвращаясь к уравнению Аррениуса, отметим, что величина e — Е* / RT (“экспонента”) равна доле активных молекул, обладающих избыточной энергией Е* для вступления в химическое взаимодействие, а коэффициент А (предэкспоненциальный множитель) равен полной частоте соударений между молекулами реагирующих веществ в реакционном объёме.
Логарифмируя уравнение Аррениуса, получим уравнение прямой, не проходящей через начало координат:
| E* ln k = ln A — ¾¾ . RT |
Построив по экспериментальным данным график зависимости ln k от 1/T (т. н. “аррениусовскую зависимость”), можно вычислить энергию активации изучаемой реакции по тангенсу угла наклона, который в этом случае равен —Е*/R (рис. 12.5).
 Рис. 12.5. Аррениусовская зависимость и энергия активации Рис. 12.5. Аррениусовская зависимость и энергия активации |
Дифференцируя логарифмическую форму уравнения Аррениуса по температуре, получим уравнение, подобное уравнению изобары (изохоры) Вант-Гоффа:
| d ln k E* ¾¾¾ =¾¾ dT RT 2 |
Интегрирование его в пределах k1 ¸ k2 и Т1 ¸ Т2 приводит к уравнению
| k2 E* 1 1 ln ¾ = ¾ (¾ — ¾) k1 R T2 T1 |
| k2 E* Т2 — Т1 ln ¾ =¾ ( ¾¾¾) , (12.11) k1 R T1 T2 |
где k1 и k2 — константы скорости данной реакции при температурах T1 и T2 соответственно.
С помощью уравнения (12.11), также называемого уравнением Аррениуса, можно вычислить константу скорости k2 при заданной температуре Т2, если известны значения константы скорости k1 при температуре Т1 и энергия активации реакции Е*. Кроме того, это уравнение позволяет вычислить энергию активации реакции по значениям двух констант скорости при различных температурах:
| R T1 T2 k2 E* = ¾¾¾¾ ln¾ . Т2 — Т1 k1 |
Таким образом, в соответствии с теорией активных соударений повышение температуры увеличивает скорость химических реакций потому, что при этом возрастает доля активных молекул, способных преодолеть потенциальный барьер при столкновении.
Теория активных соударений
Теория была сформулирована С. Аррениусом в 1889 году. В основе этой теории лежит представление о том, что для протекания химической реакции необходимо соударение между молекулами исходных веществ, а число соударений определяется интенсивностью теплового движения молекул, т.е. зависит от температуры. Но не каждое соударение молекул приводит к химическому превращению – к нему приводит лишь активное соударение.
Тот минимальный запас энергии, которым должны обладать молекулы исходных веществ для того, чтобы их соударение было активным, называют энергетическим барьером реакции. Наглядное представление об энергетическом барьере реакции дает графическое изображение энергетики химической реакции (рис. 8.4).
В качестве абсциссы в этих диаграммах используется так называемая координата реакции. Вообще говоря, она является сложной функцией межатомных расстояний. Но для практических целей и простых молекул можно считать, что она характеризует изменения в межатомных расстояниях, которые происходят при сближении исходных молекул, образующих активированный комплекс, и взаимном удалении продуктов реакции при распаде активированного комплекса. По оси ординат откладывается потенциальная энергия всей системы.
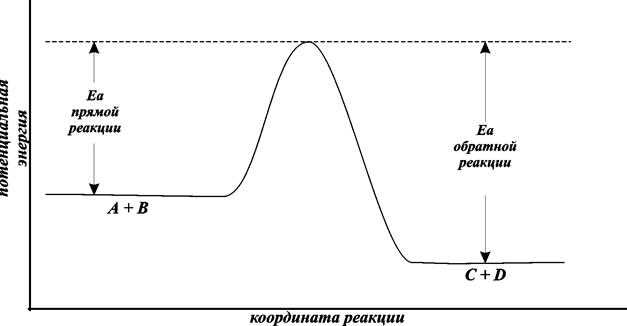
Рис. 8.4. Энергетическая диаграмма реакции А+B = C+D
То дополнительное количество энергии, которое нужно добавить к средней энергии молекул исходных веществ, чтобы соударение стало активным, называется энергией активации.
Энергия активации ощутимо влияет на значение константы скорости реакции и ее зависимости от температуры: чем больше Еа, тем меньше константа скорости и тем значительнее влияет на нее изменение температуры. Константа скорости реакции связана с энергией активации сложной зависимостью, описанной уравнением Аррениуса:
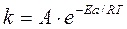 , (8.27)
, (8.27)
где А – так называемое «число соударений», которое представляет собой число соударений в одну секунду между одной молекулой вещества а и одной молекулой вещества В, заключенных в объеме в 1см 3 .
Однако наблюдаемые константы скорости реакции, как правило, гораздо меньше вычисленных по уравнению. Поэтому уравнение для константы скорости реакции видоизменяют следующим образом:
 , (8.28)
, (8.28)
где Z – теоретическое число столкновений, а Р – так называемый фактор вероятности или стерический, который учитывает все влияния, вызывающие отклонения от идеального уравнения. Необходимость ориентации может заметно тормозить даже сравнительно простые реакции. Хорошо изученным примером является реакция Н2 + J2 ® 2НJ.
Для того, чтобы простое соударение дало две молекулы йодистого водорода, надо, чтобы ориентация молекулы была сходна с той, которая изображена на рис. 8.5 а.

Рис. 8.5. Схема, отображающая значение благоприятной ориентации для того, чтобы простое соударение могло привести к образованию продукта реакции
Энергия активации этой реакции невелика, но скорость мала. Это связано с довольно жесткими требованиями, предъявляемыми к ориентации реагирующих молекул. Тогда А = РZ, т.е. а характеризует число соударений с благоприятной ориентацией и называется предэкспоненциальным множителем.
Используя уравнение Аррениуса, можно определить энергию активации Еа. Для этого уравнение Аррениуса удобно применять в логарифмической форме:
 (8.29)
(8.29)
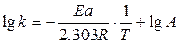 . (8.30)
. (8.30)
Если построить график lgk от 1/Е, то получим прямую, отсекающую на оси ординат отрезок, равный lgА, и имеющую тангенс угла, равный ‑ Еа/2,303R, т.е. tga = — Еа/2,303R, откуда Еа = -2,303 R tga.
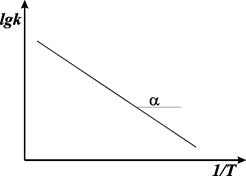
Рис. 8.6. График Аррениуса
Из уравнения видно, что константа скорости реакции k является произведением двух сомножителей. Предэкспоненциальный множитель А практически не зависит от температуры, так как последняя не влияет на взаимную ориентацию молекул. Экспоненциальный множитель e -Ea/RT , который характеризует долю активных соударений от общего числа двойных соударений, сильно зависит от температуры.
Теория активных соударений внесла в химическую кинетику новые представления об активных соударениях и об энергии активации, но эта теория не рассматривала механизм самого соударения, что является ее недостатком.
8.15.2. Теория активированного (переходного) комплекса
(переходного состояния)
Эта теория – простейший и исторически первый вариант статистической теории химических реакций. Она разработана Э. Вагнером, М. Поляни, Г. Эйрнингом и М. Эвансом в 30-х годах XX века.
8.15.3. Вывод основного уравнения теории
переходного состояния
В основу теории также положено представление о столкновении молекул как непременном условии реакции. Но она рассматривает то, что происходит в момент столкновения.
Для реакции А + В = С в соответствии с теорией переходного состояния следует: А + В « х * ® С, где х * – переходной комплекс (пк). Что подразумевается под переходным комплексом? После столкновения молекул А и В начинается перераспределение химических связей и образование переходного комплекса. переходный комплекс представляет такое состояние взаимодействующих молекул, когда старые связи еще не полностью разорвались, а новые еще не полностью сформировались. В результате мы имеем состояние промежуточное между А, В и С. Переходное состояние характеризуется непрерывным изменением расстояний между взаимодействующими атомами. В этом существенное отличие переходного комплекса от обычной молекулы, в которой средние расстояния между атомами не зависят от времени. Переходный комплекс также не следует путать с промежуточными веществами. Он является динамической структурой, для образования которой требуется затрата энергии.Энергия, необходимая для перевода реагирующих молекул в состояние переходного комплекса, носит название энергии активации. Так как исходные молекулы еще не распались, то энергия перехода в активированное состояние меньше энергии разрыва связей в молекулах исходных веществ:
В этом случае kхимического равновесия переходного комплекса равна:
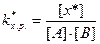 , (8.31)
, (8.31)
а концентрация х составит:
 . (8.32)
. (8.32)
Затем ПК распадается необратимо с образованием продукта С. Характеристикой (количественной) распада будет частота распада ПК – Р. Из статистической механики известно, что Р зависит только от температуры. Эта зависимость имеет вид Р = kТ/h, где k – постоянная Больцмана; h – постоянная Планка; Т – абсолютная температура.
Значит, для данной температуры число Р одинаково для всех переходных состояний, а скорость любой химической реакции определяется лишь концентрацией ПК:
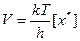 . (8.33)
. (8.33)
Концентрация ПК связана с концентрацией реагентов и поэтому, подставив их выражения, получим:
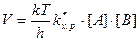 . (8.34)
. (8.34)
К элементарной реакции взаимодействия применим закон действия масс:
символ kV употребляется для константы скорости в отличие от константы Больцмана.
Приравняем правые части уравнений и получим:
 (8.36)
(8.36)
 . (8.37)
. (8.37)
Из уравнения видно, что при данной температуре константа скорости реакции зависит от константы химического равновесия образования ПК и от частоты распада ПК. Уравнение называется основным уравнением теории переходного состояния.
8.15.4. Термодинамическая форма основного уравнения теории
переходного состояния
Термодинамические данные позволяют связать 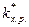 со стандартной свободной энергией образования переходного комплекса DG ¹ , называемой также свободной энергией активации:
со стандартной свободной энергией образования переходного комплекса DG ¹ , называемой также свободной энергией активации:
DG’ ¹ = -RT ln , (8.38)
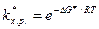 . (8.39)
. (8.39)
Тогда для константы скорости химической реакции можно записать
 . (8.40)
. (8.40)
Из этого уравнения видно, что скорость реакции определяется свободной энергией активации DG ¹ , а не Еа, как это следует из теории активных соударений Аррениуса.
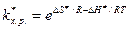 (8.41)
(8.41)
или, по теории переходного состояния, kv равна:
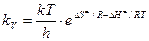 (8.42)
(8.42)
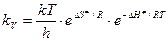 , (8.43)
, (8.43)
т.е. мы получили термодинамическую форму основного уравнения теории переходного состояния.
8.15.5. Сравнение термодинамической формы
основного уравнения теории переходного состояния
с уравнением Аррениуса
При сравнении уравнения для ТПС с уравнением Аррениуса обнаруживается, что
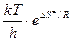 (8.44)
(8.44)
и соответствует А в уравнении Аррениуса, т.е.
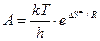 . (8.45)
. (8.45)
Значит определив опытным путем А, можно рассчитать энтропию активации DS ¹ , которая дает важную информацию о механизме образования активированного комплекса. Например, чем более жестко связывается фермент с субстратом, т.е. образуется большее число связей, тем больше уменьшается DS ¹ . Величина DН ¹ практически равна энергии активации.
Поскольку А=РZ, то:
 . (8.46)
. (8.46)
Так как Z – теоретическое число столкновений и kT/h практически постоянны, вероятностный фактор Р будет, очевидно, связан с энтропией активации DS ¹ . Если она велика и положительна, то фактор Р велик, и реакция будет быстрой. Но если DS ¹ отрицательна и мала, то реакция будет медленной, т.е. чем больше возрастание энтропии, тем больше вероятность переходного состояния.
Поскольку kT/h соответствует Z и равно при 298К 6,3× 10 -12 сек -1 , то это число равно числу активных комплексов, разлагающихся за 1 сек. в 1 см 3 или количеству соударений в 1 сек. в 1 см 3 .
Таким образом, скорость реакции, согласно теории переходного состояния, зависит от двух факторов:
1) энергетического фактора – DН ¹ , энтальпии активации (чем больше энтальпия активации, тем меньше скорость реакции);
2) энтропийного фактора – DS ¹ , энтропии активации (чем больше энтропия активации, тем больше скорость реакции).
Учет энтропийного фактора для кинетики реакций во многих отношениях оказался плодотворным и впервые позволил установить связь константы скорости со строением молекул реагирующих веществ. При этом теория ПС оперирует, в частности, величинами расстояний между атомами в молекулах, взаимной ориентацией молекул, т.е. параметрами геометрического характера.
Теория активных соударений позволяет при знании энергии активации рассчитать общее число эффективных соударений и отсюда скорость реакции, не объясняя механизма реакции. В отличие от теории активных соударений теория ПК сопоставляет различные возможные комплексы, выявляет большую или меньшую их достижимость и определяет в результате энергетически наиболее выгодный путь реакции.
Для вычисления скоростей взаимодействия двух атомов две теории дают одинаковые результаты. В случае нелинейных многоатомных молекул теория ПК дает значение скоростей, отличных от значений, которые дает теория соударений. Если известна конфигурация реагирующих молекул и активного комплекса, теория ПК позволяет рассчитать предэкспоненциальный множитель. К сожалению, в большинстве случаев строение активированного комплекса и его свойства неизвестны и это затрудняет расчеты.
Таким образом, две теории дополняют друг друга. Теория ПК применяется для вычисления абсолютных скоростей электродных процессов, процессов диффузии и т.д. Теория активных соударений хорошо описывает, как правило, реакции в газовой среде.
Лекция 9.
Основы кинетики и механизма
ферментативных реакций.
Гомогенный и гетерогенный катализ
9.1. Понятие катализа
Катализ (от греч. katalysis – разрушение) – изменение скорости химической реакции под влиянием катализаторов. Обычно под катализом понимают ускорение реакции (положительный катализ), однако в отдельных случаях подразумевается замедление реакций (отрицательный катализ). Реже приходится иметь дело с явлением автокатализа, когда катализатором служит один из продуктов реакции.
В зависимости от того, находится ли катализатор в той же фазе, что и реагирующие вещества, или образует самостоятельную фазу, говорят о гомогенном или гетерогенном катализе. В последнем случае ускорение процесса обычно связано с каталитическим действием поверхности твердого тела (катализатора). В гетерогенном катализе применяются переходные металлы, их оксиды, сульфиды и другие соединения. Гомогенными катализаторами обычно служат растворы кислот, оснований, солей и, прежде всего, солей d-элементов (Cr, Mn, Fe, Co, Ni, Cu и др.)
В ходе каталитической реакции катализатор остается химически неизменным, а его масса – постоянной (если не считать потерь за счет механического уноса и возможности протекания побочных химических процессов, в которых катализатор участвует как реагент). Между количествами реагентов и катализаторов существует огромная диспропорция. Так, одна массовая часть катализатора вызывает превращение миллиона массовых частей NH3 при его окислении в азотную кислоту.
Катализаторы отличаются избирательностью (селективностью) действия. Так, на оксиде алюминия при 350-360 °С происходит дегидратация этанола:
а в присутствии меди при 200-250 °С – его дегидрирование:
В отсутствие катализатора обе реакции идут параллельно.
Катализатор не влияет на истинное равновесие, т.е. не меняет константу равновесия и равновесные концентрации. Он в равной мере ускоряет и прямую и обратную реакции. Если повышение температуры не только ускоряет процесс, но и смещает равновесие, то катализатор лишь изменяет время его достижения. Оно тем меньше, чем активнее катализатор. Вводя катализатор в реакционную зону эндотермических реакций, можно осуществить снижение температуры, не проигрывая в скорости процесса.
Присутствие в зоне реакции посторонних веществ оказывает различное влияние на катализатор: одни нейтральны, другие усиливают действие катализатора, третьи его ослабляют и вообще прекращают. Ускорители каталитических процессов называются промоторами, или активаторами. Вещества, снижающие скорость реакции, называются ингибиторами.
9.2. Основы теории гомогенного катализа
Большинство гомогенных каталитических процессов объясняет теория промежуточных соединений. Исходное положение этой теории – предположение, что в течение реакции образуются неустойчивые, промежуточные соединения катализатора с реагирующими веществами, которые затем распадаются с образованием продуктов реакции, а катализатор регенерируется.
Так, для гипотетической реакции А + В ® С механизм протекания таков: на первой стадии образуется неустойчивое промежуточное соединение одного из реагентов с катализатором: А + К® [АК]. Затем это соединение взаимодействует с другим исходным веществом, в результате чего образуется продукт реакции, а катализатор освобождается: [AK] + В ® С + К.
Хотя участие катализатора в механизме реакции и удлиняет путь процесса, скорость его значительно увеличивается, так как энергетические затраты на образование и разрушение промежуточных соединений гораздо меньше, чем непосредственное, прямое образование продуктов реакции, – иными словами, влияние катализатора на скорость химического процесса связано с понижением энергии активации реакции.
При гомогенном катализе важное значение имеет кратковременное сочетание молекул и ионов, в результате чего образуются соединения типа диполей или содержащие водородную связь. Эти реакции могут катализироваться кислотами или основаниями. Согласно протонной теории кислот и оснований, кислота – это соединение, способное отщеплять протон, основание – вещество, способное присоединять протон. В ходе катализа происходит перераспределение электронов в молекуле субстрата, возникают промежуточные соединения с повышенной активностью (карбониевые ионы, карбанионы, полярные комплексы). При этом снижается энергия активации и ускоряется реакция.
Кислотно-основной катализ обязательно включает стадию переноса протона от одной молекулы к другой. В реакционной среде должны быть доноры и акцепторы. Если кислоту обозначить НА, субстрат – НХ, а В и А — – основания, НХН + и Х — – ионизированные формы субстрата, ХН – продукты реакции, то катализ кислот можно записать следующим образом:
НХ + на « НХН + + А —
В + НХН + « ВН + + ХН
Гомогенных каталитических реакций в растворах, ускоряемых ионами гидроксила и водорода, довольно много. К этому типу относятся реакции этерификации, инверсии сахаров, омыление сложных эфиров и т.д.
Омыление сложных эфиров катализируется как кислотой, так и основанием. В последнем случае оно протекает по уравнению 2-го порядка и может быть записано:
9.3. Основы теории гетерогенного катализа
Одно из современных объяснений механизма действия твердых катализаторов дает мультиплетная теория. Она была предложена в 1929 году А.А. Баландиным. В основу мультиплетной теории положены принципы структурного и энергетического соответствия между поверхностью катализатора и молекулами реагирующих веществ. Структурное соответствие заключается в том, что расстояние между атомами кристаллической решетки катализатора и валентные углы между ними должны соответствовать этим же параметрам участвующих в реакции веществ. В таком случае молекулы реагирующих веществ совершают активированную адсорбцию на катализаторе, которая состоит в том, что образуется множество связей между атомами адсорбента (катализатора) и адсорбтива (реагирующего вещества). При этом происходит «ослабление» внутримолекулярных связей реагирующих веществ – активация.
Например, расстояние между атомами Ni – Ni на поверхности никеля Ренея наилучшим образом подходит к ацетиленовой связи, а к этиленовой связи в равной мере подойдут расстояния между атомами многих металлов – от платины до железа. Вот поэтому никель Ренея обладает уникальной селективностью по отношению к реакциям гидрирования ацетиленовых углеводородов, а металлы платиновой группы используются в процессах получения соединений этилена.
Эту теорию можно было бы назвать теорией геометрического подобия активного центра и реагирующей молекулы. Для различных реакций число адсорбционных центров (каждый из которых отождествляется с одним атомом металла) в активном центре принимается равным 2, 3, 4, 6 и т.д. Подобные активные центры были названы дуплетами, триплетами, квадриплетами, секстетами, а в общем случае мультиплетами.
Поверхностное соединение, образующееся в результате активированной адсорбции молекул реагирующих веществ на отдельном мультиплете, называют мультиплетным комплексом. Он отображает физическое состояние вещества в момент его превращения (исходные вещества прекратили свое существование, а продукты реакции еще не образовались). Мультиплетный комплекс распадается с образованием или продуктов реакции, или исходных веществ. Этим управляют законы химического равновесия.
Согласно другому принципу мультиплетной теории – энергетическому соответствию – наибольшая эфективность твердого катализатора достигается при определенной энергии мультиплетного комплекса, соответствующей равенству поверхностных активностей исходных веществ и продуктов реакции по отношению к данному катализатору.
Энергетическое соответствие катализатора определяют экспериментально, поэтому мультиплетная теория не в состоянии однозначно предсказать катализатор той или иной реакции.
В отличие от мультиплетной теории, теория каталитически активных ансамблей Н.И. Кобозева предусматривает возможность существования активных центров из атомов, не входящих в кристаллическую решетку. Из этой теории следует, что лишь сочетание определенного (обычно небольшого) числа частиц катализатора (ансамбль) способно проявлять «катализаторскую» активность.
Так, для реакции соединения азота и водорода необходимо три атома катализатора (железа), сгруппированных в активный ансамбль. Для реакции присоединения водорода к органическим соединениям, ускоряемой палладием, необходимо два атома палладия и т.д. Отдельные ансамбли на поверхности твердого катализатора не могут соединяться друг с другом, потому что поверхность катализатора очень неоднородна, и частицы вещества не могут беспрепятственно двигаться по всей поверхности. Их движение ограничено лишь небольшой частью поверхности. Согласно теории каталитически активных ансамблей, атомы активных центров образуют «аморфную» фазу на поверхности кристаллических граней самого металла. Последний выполняет при этом роль носителя.
9.4. Ферменты как биологические катализаторы
Химические процессы в живых организмах осуществляются при помощи биологических катализаторов – ферментов. Все известные в настоящее время ферменты являются белками, многие из которых содержат ионы металлов.
По эффективности ферменты значительно превосходят химические катализаторы. Во многом это обусловлено тем, что ферменты резко снижают энергетические барьеры на реакционном пути. Например, энергия активации для реакции распада перекиси водорода под действием иона железа (II) и молекулы каталазы соответственно 42 и 7,1 кДж/моль, для гидролиза мочевины кислотой и уреазой – 103 и 28 кДж/моль соответственно.
Кроме того, ферменты отличает высокая специфичность и направленность действия. Так, амилаза, содержащаяся в слюне, легко и быстро расщепляет крахмал, молекула которого состоит из большого числа одинаковых глюкозных звеньев. Но она не катализирует процесс распада сахарозы.
Каталитические действия ферментов происходят в сравнительно «мягких» условиях (t = 37–40 o C, при невысоком давлении и определенном значении рН).
9.5. Кинетика реакций, катализируемых ферментами
Уже в ранних исследованиях по влиянию концентрации реагента на скорость ферментативных реакций была обнаружена очень важная особенность ферментативного катализа, которая заключается в сложном характере кинетики этих реакций. Так, при низких концентрациях реагента (или субстрата) реакция протекает в соответствии с уравнением первого порядка. При высоких концентрациях субстрата скорость перестает зависеть от концентрации и, таким образом, реакция в этих условиях протекает в соответствии с уравнением нулевого порядка. Общий вид зависимости скорости реакции от концентрации субстрата при такой двухступенчатой кинетике приведен на рис. 9.1.

Рис. 9.1. Зависимость скорости реакции, катализируемой ферментом, от концентрации субстрата
Л. Анри первым предположил, что фермент образует промежуточное соединение или комплекс с субстратом. При высоких концентрациях субстрата весь фермент будет в комплексе с субстратом и скорость реакции будет максимальной. Иными словами, в реакциях, катализируемых ферментами, стадией, определяющей скорость процесса, будет стадия распада комплекса на фермент и продукт. Она носит название лимитирующей стадии всего процесса.
Обозначим фермент Е, а субстрат S и запишем уравнение ферментативной реакции.
 (быстрая стадия). (9.1)
(быстрая стадия). (9.1)
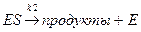 (медленная стадия), (9.2)
(медленная стадия), (9.2)
Такие реакции как в химической, так и ферментативной кинетике носят название сложных. Их можно решать как равновесным, так и методом стационарного состояния.
Равновесный метод дает хорошие результаты, если состояние, близкое к равновесному, устанавливается на быстрой стадии. Именно этим методом Михаэлис и Ментен провели математическую обработку экспериментальных данных, подобных представленным на рис. 9.1. Сложность решения заключается в том, что необходимо выразить концентрацию равновесную [ES] через величины, которые можно измерить количественно. Это позволяет исключить [ES] из уравнения скорости реакции. Константа равновесия первой стадии может быть представлена в виде
 . (9.4)
. (9.4)
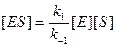 . (9.5)
. (9.5)
На этом этапе задача еще не решена, потому что [E] – это концентрация свободного фермента, которую также трудно измерить экспериментально. Однако в состоянии равновесия содержание Есвоб. и Есвяз. можно выразить через Еt, где Е t – общая концентрация фермента:
Это уравнение носит название уравнения постоянства общей концентрации фермента. Общая концентрация фермента может быть измерена. Множитель в уравнении (9.5) можно заменить на разность [Et] – [ES]. В результате уравнение примет вид
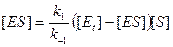 . (9.7)
. (9.7)
Решая его относительно [ES], получим:
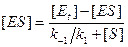 . (9.8)
. (9.8)
Вместо k-1/k1 введем КМ. Новая константа по своей сути является константой диссоциации фермент/субстратного комплекса ES, в то время как К, входящая в уравнение (9.4), является константой его образования. Новую константу, численно равную отношению k-1/k1, называют константой Михаэлиса и обозначают обычно как Км. Тогда уравнение (9.8) принимает вид
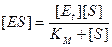 . (9.9)
. (9.9)
Теперь мы имеем выражение, которое можно подставить в общее уравнение скорости: V = k2 [ES] и получим
 . (9.10)
. (9.10)
Сделаем еще одно упрощение: когда весь фермент связывается с субстратом (при высокой концентрации субстрата), скорость становится максимальной. Свободного фермента уже нет и [Et] = [ES]. В этих условиях скорость реакции обозначают Vmax. Величина Vmax является константой, характеризующей реакцию в условиях, когда она протекает по уравнению нулевого порядка. Это уравнение имеет вид
и тогда уравнение (9.10) примет вид
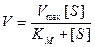 .
.
В результате такой подстановки мы имеем уравнение Михаэлиса-Ментен, позволяющее легко измерять максимальную скорость из экспериментальных данных, тогда как величины Et и k2 определять сложнее.
Конечно, сегодня имеется ряд очищенных ферментов с известным молекулярным весом и для них можно рассчитать молярную концентрацию Et. Но для всех неочищенных препаратов, а также для ферментов с неизвестным молекулярным весом применение Vmax вместо k2[Et] существенно упрощает расчеты.
Действительно, по этому уравнению, если концентрация S мала по сравнению с КМ, то член [S] можно исключить из знаменателя, и уравнение Михаэлиса-Ментен приобретает вид
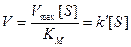 (первый порядок).
(первый порядок).
При высоких концентрациях субстрата можно считать, что величина [S] значительно больше КМ. При этом
V = Vmax = const (нулевой порядок).
Считается, что при изучении многих реакций, катализируемых ферментами, лучше пользоваться не равновесным методом, а методом стационарного состояния. Рассмотрим, как в этом случае будут различаться окончательные уравнения скорости.
Начальные стадии в обоих вариантах одинаковы:
;
;
После достижения стационарного состояния [ES] будет постоянна и, следовательно, скорость ее изменения будет равна нулю:
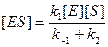 . (9.12)
. (9.12)
Различие между выражением (9.12) и (9.5) заключается в константах: равновесный метод дает член k1/k–1, а метод стационарного состояния – член k1/(k –1 + k2). Обратной величиной последнего будет новая константа, а именно константа Михаэлиса КМ в условиях стационарного состояния. Все остальные преобразования аналогичны первому случаю. Окончательная форма записи уравнения скорости в условиях стационарного состояния реакции имеет вид
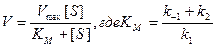 . (9.13)
. (9.13)
Это и есть уравнение скорости, которое применяется в ферментативной кинетике, если нет убедительных данных, свидетельствующих о том, что начальная стадия реакции быстро приходит к равновесию.
Поскольку уравнения (9.13) и (9.11) являются уравнениями гиперболы, графически определить постоянные величины Кm и Vmax довольно трудно, поэтому уравнение Михаэлиса-Ментен было преобразовано в другие более удобные формы. Одна из них – форма уравнения Лайнуивера-Берка. Если уравнение (9.13) записать в обычной форме, а затем обе стороны уравнения выразить в виде обратных величин, то получаем равенство
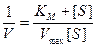 . (9.14)
. (9.14)
Правую часть уравнения можно представить так:
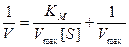 . (9.15)
. (9.15)
Две переменные V и [S] теперь разделены, и если построить график в координатах (1/V,1/[S], то мы получим прямую линию с наклоном, равным Кm/Vmax, пересекающую ось 1/V в точке 1/Vmax.
Однако для некоторых ферментативных систем график, построенный в этих координатах, может отличаться от прямой линии. Это, возможно, обусловлено тем, что при избыточных концентрациях субстрата фермент может ингибироваться или активироваться субстратом. Для аллостерических ферментов кривые насыщения субстратом обычно имеют сигмоидную форму и часто могут быть описаны уравнением, в котором [S] заменено на [S] n . Если аномалии появляются экспериментально при низких концентрациях субстрата, то соответствующие точки будут находиться только в правой части графика. Следовательно, эти аномалии не должны мешать расчету величин Km и Vmax. Если же отклонения от линейности обнаруживаются при высоких концентрациях субстрата, то соответствующие точки будут находиться рядом с точкой пересечения и возникнет проблема с экстраполяцией линейных участков полученной кривой. В таких случаях может быть использована другая форма уравнения Лайнуивера-Берка. Для этого умножим обе части уравнения (9.15) на [S] и получим:
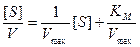 . (9.16)
. (9.16)
Построение графика в координатах [S]/V и S дает прямую линию с наклоном, равным 1/Vmax, пересекающую ось [S]/V в точке Km/Vmax. Все отклонения от графика, проявляющиеся при больших значениях [S], будут находиться в правой части графика и с ними можно не считаться.
Упомянем еще об одной часто применяемой альтернативной форме уравнения Михаэлиса-Ментен, поскольку она обладает тем преимуществом, что позволяет заметить такие отклонения от линейности, которые могли быть пропущены в случае других графиков.
Если умножить уравнение (9.15) на Vmax·V и произвести соответствующие преобразования, то получим:
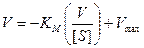 . (9.17)
. (9.17)
Отметим, что точка пересечения прямой с осью ординат соответствует Vmax, точка пересечения с осью абсцисс соответствует величине Vmax/Km, а наклон прямой равен –Km.
В настоящее время для анализа кинетических параметров ферментативных реакций все больше используется ЭВМ, что расширяет перечень подходов и делает анализ более объективным.
9.6. Физический смысл величин Km и Vmax
Как правило, для характеристики каждого фермента определяют его константу Михаэлиса, которая легко воспроизводится и не зависит от концентрации фермента. Физический смысл этой константы заключается в том, что она численно равна концентрации субстрата, при которой активность фермента составляет половину максимальной. Это легко показать с помощью небольшого алгебраического преобразования, подставив вместо V величину 0,5Vmax:
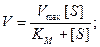
 . (9.18)
. (9.18)
Разделим (9.18) на Vmax и получим:
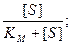
Поэтому Km имеет размерность моль/л.
Фундаментальное значение величины Vmax связано с ее отношением к величине k2. Для специального случая, когда весь фермент входит в комплекс ES, мы можем заменить величину k2[Et] на Vmax.
Если концентрация фермента выражена в моль/л, а скорость превращения субстрата – моль/л мин., то k2 равна максимальному числу оборотов фермента:
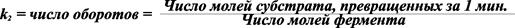 .
.
Поскольку скорость реакции зависит от температуры, число оборотов относят к какой-то конкретной температуре. Для определения числа оборотов необходимо знать молекулярный вес фермента и его концентрацию в растворе. При этом необходимо помнить, что все эти расчеты относятся к ферментам с одним активным центром. Очень часто число оборотов рассчитывают по количеству образовавшегося продукта.
9.7. Уравнение Михаэлиса-Ментен
и ферментативные механизмы
Изучение механизма реакции по уравнению Михаэлиса-Ментен не всегда свободно от недостатков. Самым явным из них является предположение о том, что существует только один промежуточный комплекс – ES. По-видимому, более близкий к истинному механизм можно записать уравнением
 . (9.19)
. (9.19)
Определение величин наклона кривой и точки пересечения по методу Лайнуивера-Берка позволяет найти константы km и Vmax. Однако если механизм достаточно сложен, то в величину km входят добавочные константы скорости реакции, и в этом случае интерпретировать ее трудно. Поэтому интерпретацию механизма ферментативной реакции лишь на основании линейности графиков Лайнуивера-Берка необходимо проводить с большой осторожностью.
Теория активных столкновений
Теория активных столкновений (С. Аррениус) основана на том, что химическое взаимодействие осуществляется только при столкновении активных частиц, которые обладают достаточной энергией для преодоления потенциального барьера реакции и ориентированы в пространстве друг относительно друга. Чтобы произошла реакция, частицы в момент столкновения должны обладать некоторым минимальным избытком энергии, называемым энергией активации.
В теории активных столкновений считается, что акт превращения начальных веществ в конечные продукты совершается в момент столкновения активных молекул и протекает мгновенно. При этом молекулы рассматриваются как бесструктурные частицы, хотя в действительности химические реакции происходят путем постепенной перестройки молекул и перераспределения энергии между химическими связями.
Согласно молекулярно-кинетической теории энергия активации равна разности между средней энергии активных столкновений и средней энергии всех столкновений. Доля активных молекул, как показывают расчеты, составляет примерно от 10 -20 до 10 -10 . Если эта доля меньше, то скорость реакции мала, если же она больше, то реакция происходит быстро, иногда практически мгновенно. Чем выше энергия активации данной реакции, тем при более высоких температурах она совершается. Энергия активации ниже энергии диссоциации реагирующих молекул, так как для протекания реакции достаточно такого ослабления связей в молекулах, при котором начинают преобладать силы образования новых связей.
Источники активации могут быть самые разнообразные. Реакции между ионами в растворе происходят с небольшой энергией активации, которая требуется для дегидратации ионов. Реакции между свободными атомами и радикалами не требуют энергии активации, так как атомы и радикалы являются активными частицами. В гомогенных газовых реакциях основным источником активации служат столкновения, доля которых определяется законом распределения Больцмана и растет с температурой. В гетерогенных каталитических реакциях источниками активации могут служить изменения, происходящие в реагирующих молекулах при адсорбции их поверхностью катализатора. Активация может быть вызвана также внешними причинами: поглощением квантов света при фотохимических реакциях, действием электрических разрядов, ударом электронов, α – частиц, нейтронов и других излучений.
При подсчете числа столкновений нужно учитывать эффективный диаметр молекул ?. Рассмотрим элементарную бимолекулярную реакцию:
А + В → Продукты (XI.1)
Предположим, что молекула А неподвижна, а молекулы В движутся в пространстве параллельно прямой, проходящей через центр молекулы А. При отсутствии взаимодействия между молекулами А и В с молекулой А столкнутся все молекулы В, центры которых находятся внутри цилиндра, имеющего радиус r.

где σ1 и σ2 – диаметры молекул А и В соответственно. При притяжении между молекулами А и В прямолинейные пути молекул В, начиная с некоторого расстояния, искривляются, и молекулы сближаются, в результате чего с молекулой А столкнется часть молекул В, центры которых первоначально находились вне цилиндра с радиусом r. Тогда

При отталкивании молекул

где σ’1, σ’2, σ»1, σ»2 – эффективные диаметры молекул. Таким образом, эффективный диаметр молекул характеризует не только диаметры сталкивающихся молекул, но и взаимодействие между ними. Величина πσ 2 12 называется сечением соударений и имеет большое значение в современной теории кинетики химических реакций.
Эффективный диаметр σ молекул одного вида в газе рассчитывается с помощью молекулярно-кинетической теории или по эмпирическим уравнениям. Средний эффективный диаметр при столкновении с молекул разного вида вычисляем по уравнению
 (XI.2)
(XI.2)
Согласно молекулярно-кинетической теории газов полное число столкновений L0 за 1 с в 1 м 3 между одинаковыми молекулами рассчитываются по уравнению
 (XI.3)
(XI.3)
где n – число молекул в 1 м 3 ; m – масса частиц, кг.
Если в системе реагируют молекулы двух разных видов, то
 (XI.4)
(XI.4)
Число столкновений активных молекул La, рассчитанное на основе закона распределения Максвелла–Больцмана, определяется соотношением
 (XI.5)
(XI.5)
где L0 – полное число столкновений; E’ – энергия активации.
Исходя из теории активных соударений и молекулярно-кинетических представлений, вычислим константу скорости элементарной бимолекулярной реакции (XI.1) с участием молекул двух видов. Скорость рассматриваемой элементарной реакции согласно основному постулату химической кинетики выражается уравнением
где k – константа скорости; c1 и c2 – концентрации веществ А и В, моль/м 3 ;
 и
и  (XI.7)
(XI.7)
где n1 и n2 – число частиц А и В в 1 м 3 ; NA – число Авогадро.
Число активных столкновений равно числу реагирующих молекул А или В:
 (XI.8)
(XI.8)
При этом скорость реакции
 (XI.9)
(XI.9)
И с учетом (XI.8) и (XI.5) примет вид
 (XI.10)
(XI.10)
Приравнивая правые части уравнений (XI.6) и (XI.10) с учетом (XI.7), получаем
 (XI.11)
(XI.11)
Подставляя L0 из (XI.4) в (XI.11), получаем уравнение для расчета константы скорости реакции
 (XI.12)
(XI.12)
 (XI.13)
(XI.13)
 (XI.14)
(XI.14)
Можно написать вместо (XI.13)
Подставляя (XI.15) в (XI.12), получаем:
 (XI.16)
(XI.16)
Логарифмирование уравнения (XI.16) дает:
 (XI.17)
(XI.17)
Дифференцирование по T равенства (XI.17) приводит к соотношению (так как В’ приближенно не зависит от температуры)
 (XI.18)
(XI.18)
Прологарифмированное уравнение Аррениуса

где k – константа скорости реакции; A – предэкспоненциальный множитель; E – энергия активации. Продифференцируем его и сравним с (XI.18), то получим
 (XI.19)
(XI.19)
При температуре 300 — 400 К RT/2 = 1,2 — 1,4 кДж/моль. Поскольку энергия активации химической реакции обычно имеет значение от 50 до 200 кДж/моль, то при практических расчетах можно считать Е ≈ Е’. Поэтому для приближенного расчета констант скоростей бимолекулярных реакций вместо Е’ можно использовать энергию активации Е, вычисленную по уравнению Аррениуса на основании опытных данных.
Уравнение (XI.12) можно рассматривать как теоретическое обоснование уравнения Аррениуса на основе теории активных столкновений. Энергия активации в теории активных столкновений не вычисляется, а определяется опытным путем по зависимости скорости реакции от температуры. (Для некоторых сравнительно простых элементарных реакций энергия активации может быть вычислена из квантово-химических представлений). Предэкспоненциальные множители для бимолекулярных элементарных реакций рассчитываются по уравнению (XI.13). Однако бимолекулярные реакции, для которых экспериментально найденные предэкспоненциальные множители совпадают с рассчитанными, встречаются сравнительно редко. Чаще всего предэкспоненциальные множители, рассчитанные теоретически как для реакций в газах, так и растворах, значительно превышают экспериментальные значения. Это связано с упрощенным характером теории активных столкновений, которая считает, что столкновения между молекулами аналогичны столкновениям упругих шаров. В связи с этим в уравнение (XI.12) вводится множитель P, учитывающий отклонение теоретических расчетов от опытных данных. Этот множитель называется стерическим или энтропийным фактором. Уравнение (XI.12) с учетом этого фактора принимает вид
 (XI.20)
(XI.20)
При столкновении активных молекул должно быть вполне определенное расположение в пространстве активных групп, входящих в состав молекулы, которое бы обеспечило образование конечных продуктов. Стерический фактор P в большинстве случаев характеризует вероятность определенной геометрической конфигурации частиц при столкновении.
Есть и другие причины, приводящие к расхождению теории активных столкновений с опытом, которые также учитываются стерическим или энтропийным фактором. Вследствие туннельного эффекта элементарный акт может произойти при значениях энергии активации меньше Е. Это формально характеризуется величиной Р > 1. Вновь образующиеся молекулы могут быть сильно возбужденными. Если такие молекулы не освободятся от избытка энергии после своего возникновения, то они вновь могут превратиться в молекулы исходного вещества; в этом случае Р > k2
 (XI.94)
(XI.94)
где k1 = cAB / (cA cB) – константа равновесия. Вторая стадия реакции является лимитирующей.
Во втором предельном случае при k-1 3 раствора; dc2 / dr – градиент концентрации вещества B.
Чтобы определить поток J , разделим переменные r и c2 и проинтегрируем (XI.96) от R * до ∞ и от 0 до c2:
 (XI.97)
(XI.97)
где R * – расстояние между молекулами A и B при образовании пары столкновения; полагаем, что на расстоянии меньше R, c2 = 0.
Для простоты полагаем, что раствор достаточно разбавленный, и в процессе диффузии молекул B к молекуле A через слой раствора они не встречаются с другими молекулами, A и, поэтому, поток J имеет постоянное значение.
В результате интегрирования (XI.97), получаем

Скорость реакции, контролируемая диффузией, определяется скоростью потока молекул B к молекуле A. При этом скорость реакции w равна числу пар столкновения, которые образуются в 1 с в 1 м 3 раствора:
 (XI.99)
(XI.99)
где n1 = NA c1 – число молекул A в 1 м 3 раствора.
Подставляя значение J из (XI.98) в (XI.99), определяем
где D = D1 + D2, так как нужно учесть, что молекулы A также диффундируют в растворе навстречу молекулам B.
Приравнивая правые части уравнений (XI.100) и (XI.92), получаем для k3 выражение
Это выражение можно преобразовать, если для D1 и D2 использовать соотношение Стокса–Эйнштейна
 и
и  (XI.102)
(XI.102)
 (XI.103)
(XI.103)
где η – коэффициент вязкости раствора (или растворителя для достаточно разбавленного раствора). Подставляя (XI.103) в (XI.101), получаем
 ;
;  (XI.104)
(XI.104)
Таким образом, в рассмотренном предельном случае ( k1 ≠ → Продукты (XI.105)
константа скорости согласно (*) (при χ = 1) равна
 (XI.106)
(XI.106)
Но термодинамическая константа равновесия в растворе выражается через активности
 (XI.107)
(XI.107)
 (XI.108)
(XI.108)
Подставляя уравнение (XI.108) в (XI.106), получаем
 (XI.109)
(XI.109)
При γA = γB = γ ≠ = 1 уравнение (XI.109) преобразуется к виду

где k0 – константа скорости реакции, коэффициенты активности равны единице. Между константами k и k0 имеется соотношение
 (XI.110)
(XI.110)
С этим случаем мы встречаемся, например, когда одна и та же реакция может проводиться как в газовой фазе ( k0 ), так и в растворе ( k ).
Если исходные вещества A и B являются молекулами и, кроме того, принять γA = γB = γ ≠ = γ, то из выражения (XI.110) получаем, что k = k0 γ. Это означает, что для бимолекулярных реакций между молекулами константы скорости реакции при проведении ее в газовой фазе и в растворе различны.
Для мономолекулярной реакции
А → А ≠ → Продукты (XI.111)
Рассуждая аналогично, получаем выражение
 (XI.112)
(XI.112)
Если вещество A находится в растворе в молекулярной форме и принимая γA = γ ≠ = γ, то из уравнения (XI.112) получаем, что k = k0, т.е. для мономолекулярной реакции теория абсолютных скоростей реакций предсказывает слабое влияние растворителя и его природы на кинетику реакции, если конфигурация активированного комплекса мало отличается от исходных молекул.
Опыт во многих случаях подтверждает теорию. Например, мономолекулярная реакция разложения оксида азота ( N2O5 ) в газовой фазе при температуре 293 К имеет константу скорости, равную 3,4 ⋅ 10 -5 с -1 . При использовании в качестве растворителя хлороформа, дихлорэтана, нитрометана, жидкого брома и тетрахлорида углерода константы скорости при той же температуре равны соответственно 3,7 ⋅ 10 -5 ; 4,2 ⋅ 10 -5 ; 3,1 ⋅ 10 -5 ; 4,1 ⋅ 10 -5 с -1 . В случае бимолекулярных элементарных реакций перенос реакции из газовой фазы в раствор, а также изменение природы растворителя, как правило, заметно влияют на константу скорости реакции в соответствии с предсказанием теории абсолютных скоростей реакций.
Важное подтверждение теории абсолютных скоростей реакций получила для реакций между ионами в растворах сильных электролитов, так как в этом случае коэффициенты активности могут быть вычислены из теории сильных электролитов Дебая–Хюккеля. Если раствор электролита разбавленный, то коэффициенты активности можно выразить приближенно с помощью предельного закона Дебая–Хюккеля:
 (XI.113)
(XI.113)
где A – теоретический коэффициент, который для водных растворов равен 0,509; zi – заряд i-го иона; I – ионная сила раствора.
Если в реакции (XI.105) исходные вещества A и B являются ионами с зарядами zA и zB , то коэффициенты активности в уравнении (XI.110) можно приближенно выразить из уравнения (XI.113), принимая, что заряд активированного комплекса равен алгебраической сумме zA + zB зарядов реагирующих ионов:


 (XI.114)
(XI.114)

Таким образом, из теории активированного комплекса следует, что если в бимолекулярной реакции в растворе участвуют два иона с одинаковыми зарядами ( zA zB > 0 ), то lg ( k / k0 ) > 0 и константа скорости реакции увеличивается с ростом ионной силы раствора. Если же заряды ионов противоположные ( zA zB < 0 ), то и константа скорости реакции уменьшается с ростом ионной силы. Кроме того, согласно уравнению (XI.114) график в координатах lg ( k / k0 ) — √I в достаточно разбавленных растворах должен изображаться прямыми линиями, выходящими из начала координат, причем чем больше произведение zA zB, тем больше должен быть угол наклона этих прямых. Опытные данные хорошо подтверждают предсказания теории (рис.2).
Влияние ионной силы раствора на константу скорости реакции между ионами из-за изменения коэффициента активности ионов в растворах сильных электролитов называется первичным (или кинетическим) солевым эффектом. В реакциях с участием одного из ионов слабого электролита (например, иона водорода слабой кислоты) посторонний электролиз может влиять непосредственно на его концентрацию и, следовательно, на скорость реакции. Это – вторичный солевой эффект.