Окисление. Бензольное кольцо, в силу своей особой стабильности, устойчиво к действию большинства окислителей. Однако, алкильные группы, связанные с кольцом, легко окисляются под действием окислителей, таких как бихромат натрия в кислой среде, оксид хрома (VI) в уксусной кислоте или перманганат калия. В результате образуются продукты окислительной деструкции боковых цепей — ароматические карбоновые кислоты:
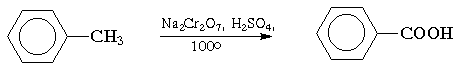
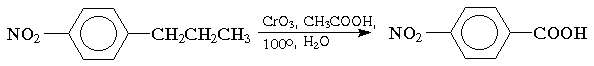
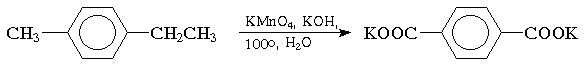
При окислении трехокисью хрома в уксусном ангидриде происходит окисление метильной группы алкиларенов до альдегидной; дальнейшему окислению до кислоты препятствует образование диацетата, который устойчив в этих условиях. Катализируемый кислотой гидролиз в водном спирте приводит к ароматическому альдегиду:
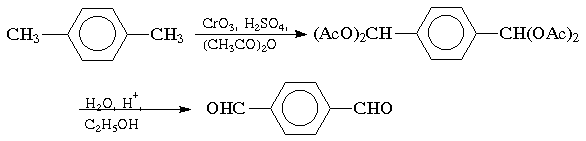
Бензиловые спирты гладко окисляются до альдегидов при применении в качестве окислителя свежеосажденной двуокиси марганца:

Окисление конденсированных ароматических углеводородов приводит к различным продуктам в зависимости от используемого реагента и условий реакции. Реагенты на основе хрома (VI) в кислой среде окисляют нафталин и алкилнафталины до нафтохинонов, тогда как бихромат натрия в водном растворе окисляет только алкильные группы. Окисление нафталина перманганатом калия в щелочной среде сопровождается деструкцией одного ароматического кольца с образованием моноциклических дикарбоновых кислот: 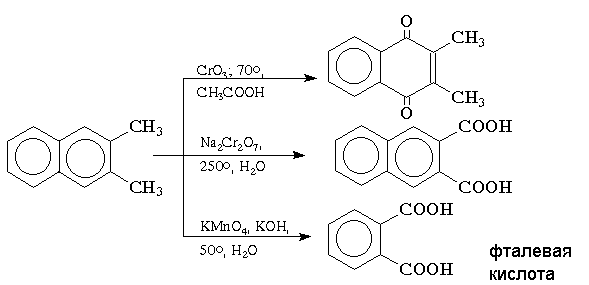
Антрацен гладко окисляется бихроматом натрия в серной кислоте или оксидом хрома (VI) в уксусной кислоте до антрахинона:
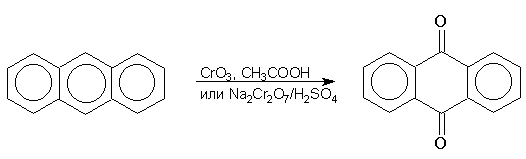
Гидрирование. Хотя ароматическое кольцо бензола гидрируется в значительно более жестких уловиях, чем двойная или тройная связь алкенов и алкинов, бензол и его производные могут быть прогидрированы до производных циклогексана над никелем Ренея (Т 120-150 о и давление 100-150 атм). Более эффективны катализаторы платиновой группы, среди которых лучшими являются родий или рутений, нанесенные на окись алюминия.
При гидрировании диалкилбензолов на Rh или Ru обычно образуется в основном цис-изомер. Гидрирование на никеле Ренея не отличается стереоселективностью, всегда образуется смесь цис-, транс-изомеров. Каталитическое гидрирование бензольного кольца невозможно остановить на первой или второй стадии, поскольку циклогексадиены и циклогексены гидрируются с большей скоростью, чем ароматические соединения.
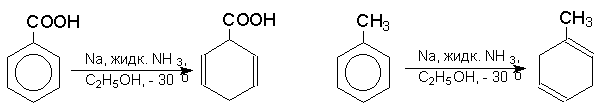
Восстановление по Берчу. Ароматическое кольцо аренов может быть восстановлено раствором натрия в жидком аммиаке в присутствие спирта как протонирующего агента. Бензол восстанавливается при этом до несопряженного циклогексадиена-1,4: (прим.44),
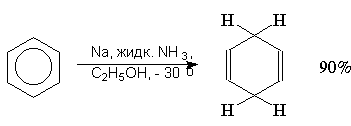
Для этой реакции предложен механизм, включающий последовательное образование анион-радикала, радикала и аниона циклогексадиена:
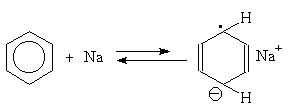
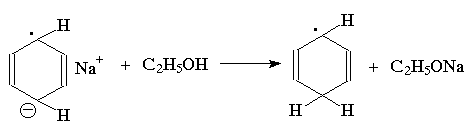
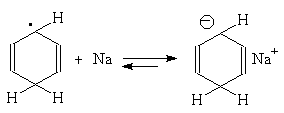
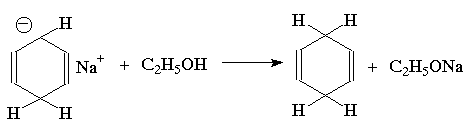
Влияние заместителей в бензольном кольце согласуется с приведенным выше механизмом: скорость восстановления возрастает при наличии электроноакцепторных заместителей и протонирование при этом происходит в положения 1- и 4- бензольного кольца; восстановление аренов с электронодонорными заместителями протекает медленнее, и протонируются положения 2- и 5-:
При восстановлении нафталина в подобных образуется 1,4-дигидронафталин:
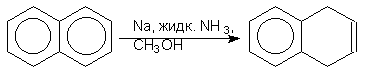
Замещенные нафталины ведут себя подобно производным бензола: при наличие в  -положении электроноакцепторной группы образуются 1,4-дигидропроизводные, а при наличии электронодонорного заместителя — 5,6-дигидропроизводные.
-положении электроноакцепторной группы образуются 1,4-дигидропроизводные, а при наличии электронодонорного заместителя — 5,6-дигидропроизводные.

Сервер создается при поддержке Российского фонда фундаментальных исследований
Не разрешается копирование материалов и размещение на других Web-сайтах
Вебдизайн: Copyright (C) И. Миняйлова и В. Миняйлов
Copyright (C) Химический факультет МГУ
Написать письмо редактору
Окисление алкенов перманганатом калия
В отличие от предельных углеводородов, алкены характеризуются высокой химической активностью, обусловленной особенностями строения молекулы. При обычных условиях алкены охотно вступают в реакции неполного окисления с превращением в органические соединения других классов. Универсальный реагент в процессах окисления алкенов – перманганат калия.
Понятие о неполном окислении
В химии органических соединений под окислением понимается взаимодействие, при котором происходит обеднение реагента водородом или обогащение кислородом, сопровождающееся отдачей электронов молекулой. Обратный процесс называется восстановлением.
Полное окисление происходит при горении углеводородов с разрушением молекулы. Продуктами в этом случае являются углекислый газ и вода. При неполном окислении продуктами становятся различные вещества.
Высокая реакционная способность алкенов обусловливается присутствием в молекуле двойной связи. Один из ее компонентов – слабая 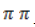 -связь – легко разрушается с образованием у углеродных атомов свободной валентности (неспаренного электрона). За счет оттягивания или отрыва освободившихся электронов и происходит окислительно-восстановительный процесс.
-связь – легко разрушается с образованием у углеродных атомов свободной валентности (неспаренного электрона). За счет оттягивания или отрыва освободившихся электронов и происходит окислительно-восстановительный процесс.

Определение степеней окисления
Для того чтобы правильно записать уравнение реакции неполного окисления алкена, нужно определить степени окисления атомов до вступления во взаимодействие и после него. Они рассчитываются исходя из электроотрицательности элементов.

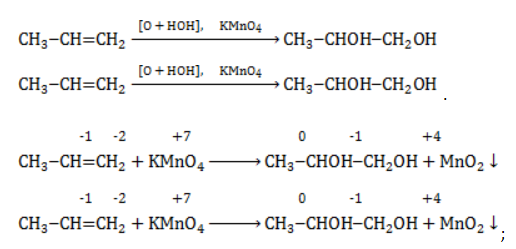
Например, при окислении пропена перманганатом калия  вступающий в реакцию пропен
вступающий в реакцию пропен  характеризуется следующими степенями окисления углеродных атомов:
характеризуется следующими степенями окисления углеродных атомов:
- В составе группы
 углерод, обладающий большей электроотрицательностью, смещает к себе электронные пары двух связей , отнимая у водородных атомов по одному отрицательному заряду. На связи сдвига электронов нет. Следовательно, атом углерода приобретает степень окисления -2 -2;
углерод, обладающий большей электроотрицательностью, смещает к себе электронные пары двух связей , отнимая у водородных атомов по одному отрицательному заряду. На связи сдвига электронов нет. Следовательно, атом углерода приобретает степень окисления -2 -2; - В группе аналогичный подсчет показывает для углерода степень окисления -1 -1 (для каждого водорода соответственно +1 +1);
- В радикале углерод оттягивает на себя отрицательные заряды с трех водородных атомов и имеет степень окисления -3 -3.
 углерод, обладающий большей электроотрицательностью, смещает к себе электронные пары двух связей
углерод, обладающий большей электроотрицательностью, смещает к себе электронные пары двух связей 



В общем виде результат можно записать следующим образом:
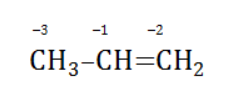
Расчет степеней окисления в кислородсодержащих соединениях производится аналогично с учетом большей электроотрицательности кислорода.

Влияние среды на окислитель
Состав раствора (наряду с температурой) определяет, до какого соединения окислится восстановитель – алкен. Окислитель в растворах с различным уровнем кислотности (щелочности) также ведет себя неодинаково.
Неорганическая соль  в водном растворе диссоциирует на катион металла
в водном растворе диссоциирует на катион металла  и собственно окислитель – перманганат-анион
и собственно окислитель – перманганат-анион  . В ходе реакции марганец восстанавливается от степени окисления +7 +7 до той или иной величины в зависимости от среды.
. В ходе реакции марганец восстанавливается от степени окисления +7 +7 до той или иной величины в зависимости от среды.
В нейтральной и слабощелочной среде марганец приобретает степень окисления +4 +4:
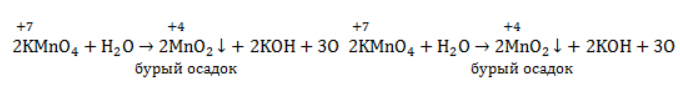
Кислород из перманганат-аниона присоединяется к алкену по месту двойной связи.
Под воздействием серной кислоты марганец восстанавливается до степени окисления +2 +2:
При окислении со щелочью (гидроксид лития достаточно высокой концентрации) марганец восстановится до +6 +6:
Мягкое окисление
Процесс в нейтральной или слабощелочной среде при обычной температуре представляет собой так называемое мягкое окисление перманганатом калия, или гидроксилирование. В алкене разрывается -связь, и к освободившимся валентностям двух углеродных атомов присоединяются две гидроксогруппы 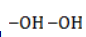 . Источниками их формирования служат:
. Источниками их формирования служат:
- кислород из перманганат-иона;
- вода.
Продукт реакции – диол (двухатомный спирт). Например, окисление этилена перманганатом калия приводит к образованию этиленгликоля:
Для составления полного уравнения нужно:
- определить степени окисления реагентов:
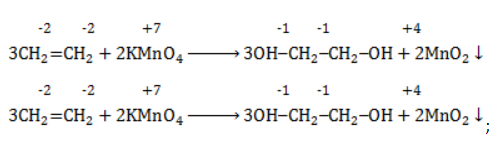
- рассчитать электронный баланс:
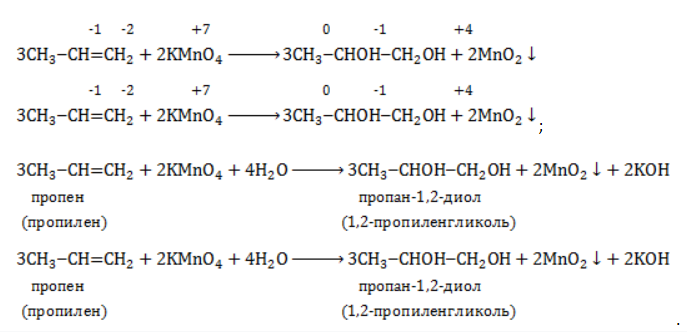
- расставить коэффициенты:
- ввести в уравнение недостающие реагенты и продукты, исходя из равенства состава в левой и правой частях уравнения, и определить окончательные коэффициенты:
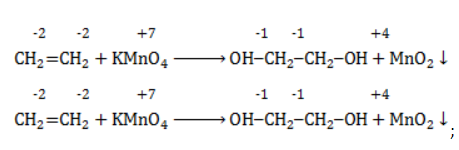

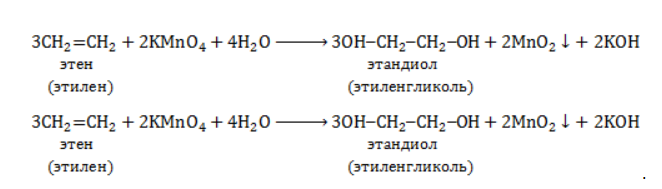
Реакция окисления пропена в нейтральной среде перманганатом калия составляется аналогично:
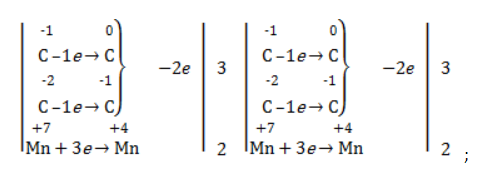
Дальше мягкое окисление не идет, так как 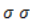 -связи в молекуле в мягких условиях сохраняются. Раствор перманганата теряет окраску, а оксид марганца выпадает в виде бурого осадка. Гидроксилирование, известное также как реакция Вагнера, служит для выявления в молекулах двойной связи.
-связи в молекуле в мягких условиях сохраняются. Раствор перманганата теряет окраску, а оксид марганца выпадает в виде бурого осадка. Гидроксилирование, известное также как реакция Вагнера, служит для выявления в молекулах двойной связи.
Жесткое окисление
Жесткими называют процессы окисления, протекающие в нейтральном растворе в условиях повышенной температуры, а также при добавлении кислоты или щелочи. В этих случаях двойная связь в алкене разрушается полностью, а продуктами реакции становятся кетоны, кислоты (с промежуточным окислением до альдегида) либо соли.

Окисление перманганатом калия в кислой среде
Пропен в содержащем кислоту растворе реагирует до образования уксусной кислоты и углекислого газа:
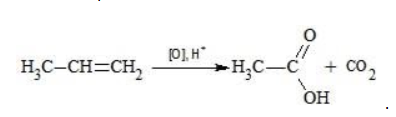
Степени окисления участвующих в реакции углеродных атомов и марганца составят:
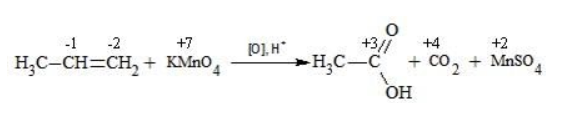
Электронный баланс определяется только с учетом углерода, вошедшего в состав кислоты:
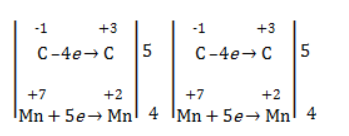
Сначала расставляются коэффициенты в окислителе, восстановителе и в продуктах окисления:
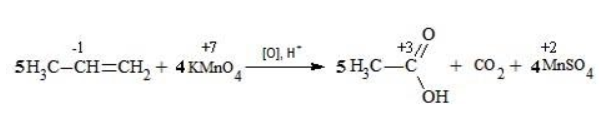
Затем вписываются недостающие вещества и полностью рассчитываются коэффициенты:

Еще один пример жесткого окисления алкенов перманганатом калия с серной кислотой – реакция с участием пентена-2. Молекула расщепляется по месту двойной связи, и ее фрагменты окисляются через промежуточное образование альдегидов до двух кислот:
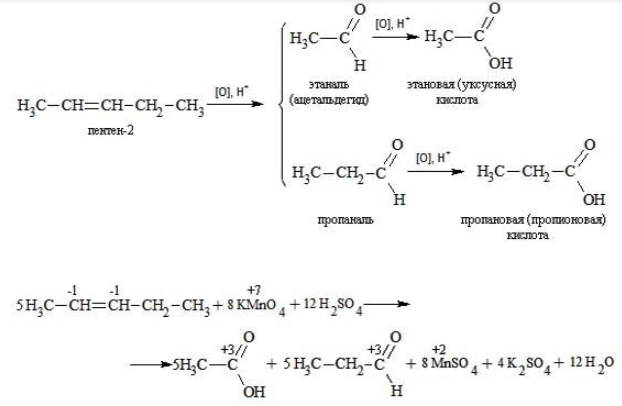
Электронный баланс составляется для двух углеродных атомов алкена, поскольку оба они являются восстановителями.
Правило, по которому осуществляется окисление углерода, отражено в таблице:

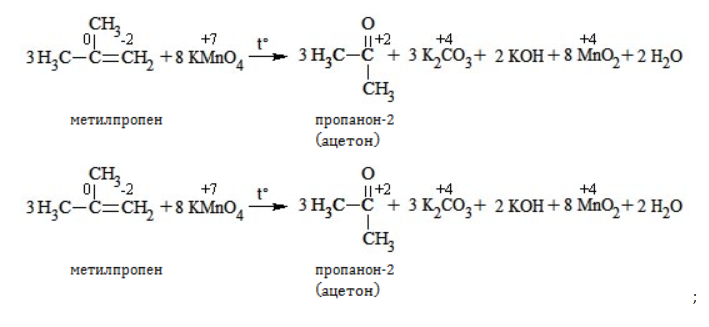
Так, в 2-метилпропене первичный атом окисляется через промежуточные формальдегид (метаналь) и муравьиную кислоту полностью – до углекислого газа, а третичный – только до ацетона:
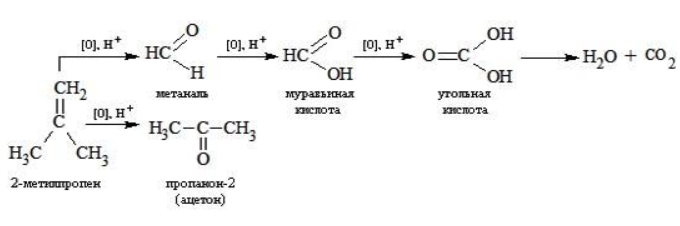
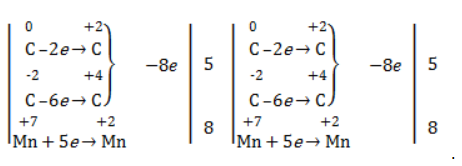
Окисление алкенов в щелочной среде
При нагревании с концентрированной щелочью алкены окисляются до солей:

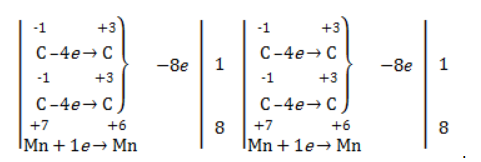
Если один из углеродных атомов – первичный, он окисляется до углекислого газа:

Окисление в нейтральном растворе
В условиях высокой температуры образующаяся щелочь вступает в реакцию, в результате которой окисление алкенов продолжается до образования кетонов или солей. Так, при жестком окислении пропена в нейтральной среде получаются те же продукты, что и в присутствии концентрированного гидроксида калия: ацетат  и неорганические соли калия – карбонат
и неорганические соли калия – карбонат  и манганат
и манганат 
 .
.
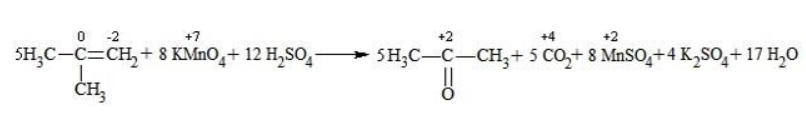
Кетон – результат окисления третичного углеродного атома, и дальнейшую реакцию они не поддерживают. Например, при окислении метилпропена как конечный продукт образуется ацетон:
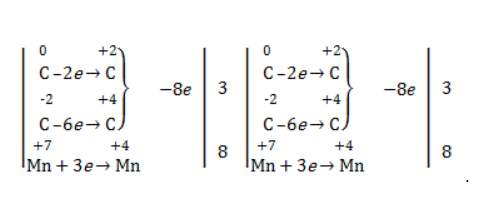
Заключение
Взаимодействие с раствором перманганата калия в мягких или жестких условиях является показателем высокой реакционной способности алкенов, которая обусловлена присутствием в молекуле легко разрываемой -связи. Реакции мягкого и жесткого окисления относятся к числу характерных химических свойств алкенов как ненасыщенных углеводородов.
Окисление (стр. 2 )
 | Из за большого объема этот материал размещен на нескольких страницах: 1 2 3 4 5 |


Эффективным гидроксилирующим агентом является тетраоксид осмия OsO4. При действии на алкены эквимолярного количества OsO4 в абсолютном эфире, пиридине или бензоле при комнатной температуре медленно образуется продукт присоединения, который при гидролизе водным раствором NaHSO3 или сероводородом дает с высокими выходами 1,2-диолы.

Тетраоксид осмия может использоваться и как катализатор в реакции окисления. Это позволяет работать лишь с незначительными его количествами, что важно вследствие высокой токсичности и стоимости данного реагента. Окисление проводят 30%-ным водным раствором пероксида водорода в трет-бутаноле при 0ºС.

Все гликоли, полученные окислением OsO4 или KMnO4, имеют цис-конфигурацию. Следует отметить, что высшие оксиды других переходных металлов (V2O5, WO3, MoO3 и др.) катализируют анти-гидроксилирование алкенов. Продукты антигидроксилирования алкенов можно также получить гидролизом соответствующих эпоксидов, либо в одном процессе совместить стадии образования и расщепления эпоксида, если алкен обрабатывать водным 30-70%-ным пероксидом водорода в муравьиной или трифторуксусной кислоте:

Кроме того, продукты транс-присоединения к двойной связи можно получить при обработке алкена иодом и бензоатом или ацетатом серебра в безводном бензоле или CCl4 (реакция Прево). Первоначально происходит образование йодэфира, в котором иод далее замещается бензоат-ионом.

Последующий гидролиз дибензоата гликоля приводит к 1,2-диолу.
3.4. ОКИСЛЕНИЕ С РАСЩЕПЛЕНИЕМ
УГЛЕРОДНОГО СКЕЛЕТА ПО ДВОЙНОЙ СВЯЗИ
Озонолиз алкенов и последующее расщепление промежуточно образующегося озонида приводит к образованию карбонильных соединений:

При взаимодействии озона (в виде 3-8%-ного раствора в воздухе) с алкенами в инертном растворителе (этилацетате, уксусной кислоте, дихлорметане) сначала происходит электрофильное присоединение озона и образуется неустойчивый мольозонид (1,2,3-триоксолан), самопроизвольно перегруппировывающийся в более устойчивый изоозонид (1,2,4-триоксолан):

Полиены более устойчивы к действию озона, сопряженные диены реагируют с озоном, как правило, по одной двойной связи.
Озониды являются неустойчивыми взрывчатыми веществами, поэтому их обычно не выделяют, а подвергают окислительному или восстановительному расщеплению. Восстановительное расщепление осуществляется с помощью цинка и уксусной кислоты, водородом в присутствии Pd или Pt, трифенилфосфином, Na2SO3, диметилсульфидом и др. При этом продуктами реакции являются альдегиды и кетоны.


Если в качестве восстановителя при расщеплении озонида использовать NaBH4, то конечными продуктами будут первичные или вторичные спирты:

Окислительное расщепление протекает под действием пероксида водорода или надкислот. В результате в зависимости от строения исходного алкена образуется либо смесь двух кетонов, либо двух карбоновых кислот, либо карбоновой кислоты и кетона.

Расщепление углеродного скелета молекулы по месту двойных связей может осуществляться и под действием других окислителей: HNO3, KMnO4, K2Cr2O7 + H2SO4, CrO3 + AcOH.

Один из современных препаративных методов окислительной деструкции алкенов был предложен в 1955 г. Р. Лемье. В основе этого метода лежит гидроксилирование алкенов с помощью KMnO4 с последующим расщеплением вицинального гликоля периодатом натрия NaIO4 при pH 7-8. Продуктами расщепления являются кетоны и карбоновые кислоты, так как альдегиды в этих условиях окисляются до карбоновых кислот. Диоксид марганца и манганат калия вновь окисляются периодатом до перманганата, что позволяет использовать только каталитические количества KMnO4.

В другой разновидности этого метода вместо KMnO4 используют каталитические количества OsO4 или RuO4 и NaIO4, что позволяет остановить окисление на стадии альдегида. Тетраоксид осмия присоединяется к двойной связи с образованием осмата, который окисляется NaIO4 до карбонильных соединений с регенерацией тетраоксида осмия:

3.5. АЛЛИЛЬНОЕ ОКИСЛЕНИЕ АЛКЕНОВ
При обработке алкенов диоксидом селена в уксусном ангидриде или смеси последнего с уксусной кислотой образуются аллиловые спирты, которые тотчас же ацилируются уксусным ангидридом и реакция на этом заканчивается:

Окисление можно проводить эфирами надкислот (обычно трет-бутиловыми эфирами надбензойной и надуксусной кислот) в присутствии солей Cu(I).

Возможно аллильное окисление алкенов и кислородом воздуха в присутствии оксида меди (промышленный способ синтеза акролеина):

Как и в случае алканов первичным продуктом окисления здесь является гидропероксид.
При действии на пропилен кислорода и аммиака протекает так называемый окислительный аммонолиз и образуется акрилонитрил:

Хотя выход акрилонитрила в этом случае не превышает 70%, окислительный аммонолиз пропилена представляет собой самый дешевый и безопасный способ производства акрилонитрила. Катализатором этого процесса служит фосфомолибдат висмута. Аналогичным образом может быть получен бензонитрил из толуола.
В мягких условиях алкины окисляются до 1,2-дикарбонильных соединений:

В большинстве же случаев окисление сопровождается расщеплением тройной связи и образованием карбоновых кислот:

Озонолиз алкинов также протекает с расщеплением тройной углерод-углеродной связи:

Под действием кислорода воздуха в спирте, пиридине и др. растворителях или гексацианоферрата (III) калия K3[Fe(CN)6] в диметоксиэтане или ДМФА ацетилениды меди (I) окисляются в 1,3-диины (реакция Глазера):

Эглинтон предложил значительно более удобную модификацию окислительной конденсации алкинов. Алкин-1 окисляют ацетатом меди (I) в растворе пиридина при 60-70°С.
Для получения несимметричных диинов используют конденсацию галогенацетиленов с алкином-1 в присутствии солей меди (I) и первичного амина (сочетание по Кадио-Ходкевичу):

5. ОКИСЛЕНИЕ АРОМАТИЧЕСКОГО КОЛЬЦА
Ароматические системы довольно устойчивы к действию окислителей. Обычно для окисления ароматического ядра приходится или применять достаточно энергичные окислители, или предварительно активировать ядро введением электронодонорного заместителя.
5.1. ГИДРОКСИЛИРОВАНИЕ АРЕНОВ
Бензол и его алкилпроизводные подвергаются окислительному электрофильному гидроксилированию в фенолы под действием пероксида водорода и органических перкислот в суперкислых средах:

Бензол окисляется до фенола реактивом Фентона – пероксида водорода, содержащего соли Fe(II) и Fe(III). При этом в результате восстановления H2O2 одноэлектронным восстановителем – ионом железа (II) – образуется гидроксильный радикал, который далее присоединяется к бензолу с образованием гидроксилциклогексадиенильных радикалов. Окисление этих радикалов ионом Fe(III) приводит к получению фенола, а при димеризации радикалов образуется 1,1′,4,4′-тетрагидробифенил-4,4′-диол, который превращается в бифенил в результате дегидратации:

Сам бензол и алкилбензолы не удается окислить в хиноны с препаративными выходами. Фенолы и первичные амины легко вступают в эту реакцию, что связано с повышением нуклеофильности ароматического ядра под влиянием электронодонорных заместителей.
Самый удобный способ получения хинонов заключается в окислении одноатомных фенолов солью Фреми – нитрозодисульфонатом калия. Эта реакция осуществляется в исключительно мягких условиях в водном спирте или ацетоне, выход обычно превышает 90%.

Приведенный на этой схеме циклогексадиеновый интермедиат был выделен, что доказывает механизм одноэлектронного окисления фенолов солью Фреми.
Незамещенный n-бензохинон кроме того получают окислением гидрохинона или анилина. В качестве окислителей используются бромат калия, хлорат натрия, дихромат калия или натрия, диоксид свинца и др. В случае анилина реакция идет через стадию образования фенилгидроксиламина, который в кислой среде перегруппировывается в n-аминофенол и затем – в монохинонимин, гидролизующийся далее в хинон:

В случае окисления гидрохинона процесс протекает через промежуточное образование хингидрона – молекулярного соединения, в котором оба фрагмента образуют структуру сэндвичевого типа и связаны переносом заряда и водородными связями.

Ди — и полиалкил-n-бензохиноны обычно синтезируют, используя более мягкие окислители – соли Fe(III):

o-Бензохиноны вследствие неустойчивости получают окислением соответствующих пирокатехинов свежеприготовленным оксидом серебра или диоксидом свинца в инертном растворителе (часто в эфире).

Для получения хинонов, содержащих электроноакцепторные заместители, применяют более жесткие окислители, например HNO3.

Конденсированные ароматические системы окисляются до хинонов значительно легче бензола. Так нафталин окисляется в 1,4-нафтохинон CrO3 в уксусной кислоте:

Алкил — и арилнафталины легко окисляются по замещенному ароматическому кольцу:

Хорошие результаты, как и в ряду бензола, дает окисление гидрокси — и аминопроизводных:

Ядро антрацена окисляется еще легче нафталинового:

В промышленности тот же самый результат достигается при окислении кислородом в присутствии оксида ванадия (V) как катализатора. Таким способом можно получать антрахинон и фенантренхинон:

5.3. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ АРЕНОВ
В промышленности окислением бензола и нафталина кислородом воздуха в присутствии V2O5 получают соответственно малеиновый и фталевый ангидриды:

При взаимодействии бензола и алкилбензолов с озоном образуются триозониды, которые далее подвергают окислительному или воcстановительному расщеплению:

Препаративное значение имеет также окисление нафталина KMnO4, приводящее к о-формилбензойной кислоте.

В замещенных нафталинах действие окислителя направляется на кольцо с большей электронной плотностью.

5.4. ОКИСЛЕНИЕ БОКОВЫХ ЦЕПЕЙ
В АРОМАТИЧЕСКИХ СОЕДИНЕНИЯХ
Окисление алкиларенов происходит в первую очередь по бензильному атому углерода, что объясняется легкостью образования соответствующего бензильного радикала.

Для окисления боковых цепей в алкилбензолах в карбоксильную группу применяют водный раствор KMnO4 при нагревании, смесь Na2Cr2O7 и H2SO4.

Следует особо отметить, что в этих условиях любая алкильная группа, содержащая атомы водорода в a-положении по отношению к бензольному кольцу, окисляется до карбоксильной.
Если алкильная группа не содержит атомов водорода в a-положении по отношению к бензольному кольцу, такая трет-алкильная боковая группа не окисляется под действием Na2Cr2O7 или KMnO4 в кислой или нейтральной среде. Так, например, трет-бутилбензол окисляется в очень жестких условиях перманганатом калия до триметилуксусной кислоты, т. е. окисляется само бензольное кольцо:

Однако водная азотная кислота окисляет трет-алкильные группы до карбоксильных.
Дихромат натрия и перманганат калия нерастворимы в ароматических углеводородах, поэтому окисление идет в гетерогенных условиях, что часто резко снижает выход продуктов окисления. Этого недостатка лишен метод межфазного переноса реагентов. Твердый перманганат калия частично растворяется в бензоле в присутствии 18-краун-6-полиэфира вплоть до концентрации 0.06 М. Такой раствор носит название «пурпурный (малиновый) бензол» и широко используется для окисления алкилбензолов:

При использовании же в качестве катализатора межфазного переноса бромида тетрабутиламмония практически весь (95%) перманганат-ион находится в органической фазе.
При синтезе аминобензойных кислот из алкиланилинов, содержащих первичную аминогруппу, используют ацильную защиту:

Алкильную группу в алкилнафталинах можно окислить до карбоксильной, не затронув при этом нафталиновые ядра, если использовать в качестве окислителя нейтральный водный раствор дихромата натрия и проводить реакцию при высоких температурах в автоклаве:

Для окисления метилбензолов в альдегиды используют смесь хромового и уксусного ангидридов. При этом образующийся альдегид превращается в ацилаль, который далее не окисляется. Омылением этого производного серной кислотой получают сам альдегид. Орто-замещенные бензальдегиды получаются таким путем с низкими выходами.

Другой способ превращения метилзамещенных бензолов в бензальдегиды состоит в их обработке хлористым хромилом в CCl4 или CS2 (реакция Этара). Вначале окисляемое вещество образует комплекс с двумя молекулами CrO2Cl2, который выпадает в осадок. При обработке комплекса водой образуется альдегид.

Данный метод дает возможность окислять только одну метильную группу в присутствии других:

Окисление метиленового звена в алкилбензолах с образованием кетонов можно провести под действием ряда окислителей (разб. HNO3, SeO2, Na2Cr2O7 и др.). Особенно легко окисляются диарилметаны:


Окисление жирноароматических соединений можно осуществлять таким образом, чтобы окислению кислородом воздуха подвергался карбанион, который образуется при депротонировании исходной СН-кислоты в инертной апротонной среде (ТГФ, диметоксиэтане). Жирноароматических соединений с pKa ниже 33-35 можно окислить в системе КОН — 18-краун-6 — ТГФ до ароматических кислот, кетонов и триарилкарбинолов:

Окисление кумола в гидропероксид с последующим разложением его серной кислотой приводит к образованию ацетона и фенола:

Аналогично из индана и тетралина можно получить соответствующие кетоны:

Важнейшее промышленное значение имеют реакции прямого окисления о— и n—ксилолов кислородом воздуха до фталевой и терефталевой кислот соответственно в присутствии ацетата кобальта (III) в уксуснокислом растворе:

Введение ацетоксигруппы в бензильное положение алкилбензолов обычно осуществляют с помощью тетраацетата свинца. Реакцию проводят кипячением смеси реагентов в бензоле или ледяной уксусной кислоте. Процесс протекает по свободнорадикальному механизму:

Соответствующие спирты получают последующим омылением ацетатов.
5.5. ОКИСЛИТЕЛЬНОЕ СОЧЕТАНИЕ ФЕНОЛОВ
Под действием одноэлектронных окислителей (гексацианоферрата (III) калия K3[Fe(CN)6], PbO2, FeCl3, Ag2O, соли Фреми, пероксодисульфата калия K2S2O8 и др.) фенолы окисляются в производные дигидроксидифенила или гидроксидифенилового эфира.

При этом сначала образуются феноксильные радикалы, которые далее подвергаются димеризации в результате образования новых связей С–С между орто-орто, орто-пара и пара-пара-положениями исходных радикалов, а также новых С–О связей между атомом кислорода одного радикала и орто-положением другой радикальной частицы. Всего, таким образом, образуется потенциально не менее пяти различных типов димеров, которые находятся в равновесии с исходным феноксильным радикалом. Далее димеры обычно изомеризуются в производные дигидроксидифенила или гидроксидифенилового эфира.

Выходы индивидуальных продуктов при окислительном сочетании фенолов сильно зависят от условий проведения реакции и природы исходных реагентов. Данный процесс имеет большое биологическое значение при биосинтезе лигнина и ряда алкалоидов.
5.6. ОКИСЛЕНИЕ ОДНОАТОМНЫХ ФЕНОЛОВ
Фенолы окисляются в n-дифенолы обработкой щелочным раствором пероксодисульфата калия (реакция Элбса). Реакция протекает через промежуточное образование гидроксифенилсульфата калия, который затем в кислой среде гидролизуется до дифенола:


Если пара-положение в исходном феноле уже занято, то реакция протекает по орто-положению, однако с меньшим выходом.
6. ОКИСЛЕНИЕ ГАЛОГЕНПРОИЗВОДНЫХ
Первичные алкилгалогениды (хлориды, бромиды, иодиды) легко окисляются в альдегиды диметилсульфоксидом (реакция Корнблюма):

Бензилгалогениды можно окислить в альдегиды, вводя их в реакцию с избытком гексаметилентетрамина (уротропина) с последующим гидролизом промежуточно образующейся четвертичной соли (реакция Соммле):

Разновидностью этой реакции можно считать получение ароматических альдегидов из четвертичных солей, образующихся из бензилгалогенидов и пиридина. Эта соль при конденсации с n-нитрозодиметиланилином, катализируемой основанием, дает интермедиат, гидролиз которого в кислой среде приводит к альдегиду:

Бензилгалогениды можно окислить в ароматические альдегиды и с помощью солей 2-нитропропана. Ароматические галогенметильные производные алкилируют амбидентный анион 2-нитропропана по атому кислорода:

Этот метод является общим способом получения ароматических альдегидов, содержащих самые разнообразные заместители в бензольном кольце. Исключение составляют лишь n-нитробензильные производные, алкилирующие анион 2-нитропропана по атому углерода.
7. ОКИСЛЕНИЕ ОДНОАТОМНЫХ СПИРТОВ
Под действием концентрированной HNO3, щелочного раствора KMnO4 и многих других окислителей первичные спирты окисляются в карбоновые кислоты:

При окислении первичных спиртов до альдегидов дихроматом натрия и серной кислотой образующийся альдегид, кипящий, как правило, при температуре значительно ниже, чем исходный спирт, немедленно удаляют отгонкой и тем предотвращают его дальнейшее окисление. Таким путем могут быть получены только низкокипящие альдегиды.

Для получения высококипящих альдегидов в качестве окислителя применяют трет-бутиловый эфир хромовой кислоты (получается из CrO3 и трет-бутанола):

Для селективного окисления первичных спиртов до альдегидов в настоящее время лучшими реагентами являются комплекс CrO3 с двумя молями пиридина: CrO3·2С5Н5N (реагент Саррета — Коллинза) и хлорхромат пиридиния CrO3·С5Н5N·HCl (реагент Кори) в хлористом метилене. Оба реагента обеспечивают очень высокие выходы альдегидов, однако хлорхромат пиридиния имеет важное преимущество в том отношении, что этот реагент не затрагивает двойную или тройную связь и поэтому особенно эффективен для получения ненасыщенных альдегидов:

Один из промышленных способов получения формальдегида заключается в окислении метанола кислородом воздуха на серебряном катализаторе:

Аллиловые, бензиловые и пропаргиловые спирты можно окислять до альдегидов диоксидом марганца:

Этот реагент окисляет в петролейном эфире или хлористом метилене ненасыщенные спирты с одной или несколькими двойными или тройными связями без изомеризации и перегруппировки.
Однако, если окисление диоксидом марганца проводить в присутствии цианид-иона, то аллиловые спирты окисляются до карбоновых кислот или их сложных эфиров (в последнем случае спирты используются в качестве растворителей). В этом варианте окисления первоначально также образуется альдегид, но в присутствии цианид-иона он дает циангидрин, который содержит группировку аллилового спирта и поэтому способен окисляться далее под действием того же окислителя.

С высоким выходом бензиловый спирт может быть превращен в бензальдегид под действием 1-хлор — или 1-бромбензотриазола.
Для превращения вторичных спиртов в кетоны используют смесь дихромата натрия и серной кислоты.

Если в молекуле спирта содержится кратная углерод-углеродная связь, то окисление ведут при комнатной или пониженной температурах в инертных неполярных растворителях строго рассчитанным количеством CrO3 в водной серной кислоте (реагент Джонса).
Первичные спирты окисляются реагентом Джонса до карбоновых кислот:

В последнее время разработан целый ряд новых эффективных методов окисления первичных и вторичных спиртов с помощью ДМСО или комплексов ДМСО с различными электрофильными агентами. Тозилаты первичных спиртов, также как и бензилтозилаты, окисляются в альдегиды при нагревании в ДМСО в течение 10-30 мин при 120-150°С в присутствии гидрокарбоната натрия как слабого основания:

ДМСО в этой реакции выполняет роль нуклеофильного агента, который замещает тозилоксигруппу по SN2 механизму с образованием алкоксисульфониевых солей. Катион алкоксисульфония далее подвергается окислительно-восстановительному элиминированию, при этом гидрокарбонат или карбонат-ион является основанием в этой Е2-реакции элиминирования, приводящей к альдегиду и диметилсульфиду.

Однако данный метод окисления первичных спиртов в альдегиды требует проведения дополнительной стадии превращения спирта в тозилат. Одностадийное селективное окисление достигается путем превращения слабого нуклеофильного агента ДМСО в сильный электрофильный агент, который непосредственно реагирует с первичными и вторичными спиртами в очень мягких условиях при 0°С и ниже. Необходимую активацию ДМСО проводят с помощью триоксида серы, трифторуксусного ангидрида, уксусного ангидрида, N-хлорсукцинимида, N, N-дициклогексилкарбодиимида С6Н11N=C=NC6H11, бромоводорода, оксалилхлорида (COCl)2, хлористого тионила SOCl2 и др. Во всех случаях в качестве реакционноспособного интермедиата образуется активированная алкоксисульфониевая соль, которая далее подвергается внутримолекулярной окислительно-восстановительной фрагментации.
Комплекс ДМСО с SО3 образуется при взаимодействии пиридинсульфотриоксида с ДМСО:

Наиболее часто используется комбинация ДМСО с N, N-дициклогексилкарбодиимидом (метод Пфитцера — Моффата):

Диметилсульфид при взаимодействии с N-хлорсукцинимидом дает хлорсульфониевую соль, которая реагирует со спиртами уже при -30°С с образованием карбонильного соединения:

Вторичные спирты можно превратить в кетоны под действием кетонов (обычно ацетона) и алкоголятов алюминия (реакция Оппенауэра). Если в систему вводить большой избыток ацетона (50-200 молей), то равновесие сдвигается в сторону образования кетона из вторичного спирта. После установления равновесия реакционную смесь обрабатывают 30%-ной серной кислотой, отгоняют избыток ацетона и образовавшийся изопропанол и выделяют продукт окисления – кетон. В качестве акцептора водорода вместо ацетона иногда берут циклогексанон (избыток до 20 молей). Катализаторами служат трет-бутилат алюминия или изопропилат алюминия (не менее 0.25 моля на 1 моль окисляемого соединения; лучше 1-3 моля на 1 моль). Реакцию проводят в кипящем бензоле или толуоле.

Роль алкоголята алюминия сводится к тому, что он образует комплекс с ацетоном и, действуя как кислота Льюиса, увеличивает избыточный положительный заряд на карбонильном атоме углерода. Благодаря этому в реакционном комплексе, включающем окисляемый спирт, облегчается гидридный переход от атома углерода, связанного с гидроксильной группой, что в конечном счете и приводит к превращению исходного спирта в кетон.

Окисление a-гидроксикетонов (ацилоинов) в соответствующие дикарбонильные соединения легко осуществляется при действии солей меди (II):

Третичные спирты весьма устойчивы к окислению и в жестких условиях происходит окисление с разрывом углерод-углеродных связей и образование кислот и (или) кетонов, содержащих меньше углеродных атомов, чем исходный спирт. Обычно окисление проводят в кислой среде. При этом спирт сначала дегидратируется до алкена, который далее легко расщепляется:

Дегидрирование спиртов. Окисление первичных и вторичных спиртов в карбонильные соединения сводится по существу к отщеплению водорода от молекулы исходного спирта. Такое отщепление можно осуществить не только с помощью окислителей, но и используя каталитическое дегидрирование. В качестве катализаторов обычно применяют тонкодисперсные медь и серебро, а также оксид цинка, которые для придания им структурной устойчивости наносят в тонкодисперсном состоянии на пемзу, асбест или другие инертные носители.

При дегидрировании изопропанола над медно-цинковым катализатором при 450ºС или оксидом цинка при 380ºС образуется ацетон:

В случае высококипящих спиртов реакцию проводят при пониженном давлении. Непредельные спирты в обычных условиях дегидрирования превращаются в соответствующие предельные карбонильные соединения. Гидрирование кратной связи осуществляется выделяющимся в процессе дегидрирования водородом.

Простейшие двухатомные спирты при каталитическом дегидрировании образуют дикарбонильные соединения:

8. РАСЩЕПЛЕНИЕ α-ГЛИКОЛЕЙ
И РОДСТВЕННЫХ СОЕДИНЕНИЙ
Под действием иодной кислоты HIO4 и ее солей, а также тетраацетата свинца Pb(OAc)4 1,2-диолы и полиолы подвергаются окислительному расщеплению с образованием альдегидов и кетонов:

Если в исходном полиоле три или более гидроксильных групп связаны с соседними атомами углерода, то из средних атомов углерода образуются карбоновые кислоты.

При использовании тетраацетата свинца вторичные спиртовые группы в полиолах окисляются до диоксида углерода.
Для окисления 1,2-диолов, растворимых в воде или в бинарной смеси вода – ТГФ, вода – диоксан, используют HIO4 и ее соли, для окисления нерастворимых в воде диолов – тетраацетат свинца в бензоле или в уксусной кислоте. В обоих вариантах окислительной деструкции 1,2-диолов в качестве интермедиата образуются циклические эфиры иодной или свинцовой кислоты. Циклические эфиры затем подвергаются окислительно-восстановительному элиминированию с образованием карбонильных соединений и иодат-иона или ацетата свинца (II) соответственно:


Для окислительного расщепления 1,2-диолов используются и другие реагенты: соли церия (IV), ванадия (V), фенилиодозодиацетат С6H5I(OCOCH3)2, хлорохромат пиридиния и др. В некоторых случаях для окисления диолов в нейтральной среде применяют пероксид никеля NiO2 и MnO2:

Такие окислители как KMnO4, Na2Cr2O7, N-иодсукцинимид также расщепляют 1,2-гликоли, но при этом вместо альдегидов образуются карбоновые кислоты.
Аналогичное расщепление наблюдается и для других соединений, содержащих атомы кислорода или азота у соседних атомов углерода: b-аминоспирты, a-гидроксиальдегиды и a-гидроксикетоны, a-дикетоны, a-кетоальдегиды, глиоксаль.

a-Дикетоны и a-гидроксикетоны расщепляются также под действием H2O2 в щелочной среде.
9. ОКИСЛЕНИЕ АЛЬДЕГИДОВ
Окисление альдегидов протекает значительно легче, чем окисление спиртов. Превращение альдегидов в карбоновые кислоты можно осуществить действием многих окислителей: HNO3, H2O2, KMnO4, NaClO2, Ag2O, щелочным раствором иода, соединениями хрома (VI). Наилучшие результаты дает обычно реагент Джонса. В этом случае окисление проводится при 0-20°С в течение короткого промежутка времени с выходами больше 80%.

Однако реагент Джонса не обладает высокой избирательностью по отношению к другим функциональным группам, а кислая среда иногда приводит к изомеризации или деструкции. В таких случаях идеальным селективным окислителем является водно-спиртовой раствор оксида серебра или раствор AgNO3 в водном аммиаке (реагент Толленса). Этот реагент не затрагивает кратные углерод-углеродные связи, гидроксильную группу спиртов и ряд других функциональных групп: