Объединенное уравнение первого и второго начала термодинамики, для равновесного обратимого процесса может быть выражено:
TdS = dU + pdV (4 – 1)
Отсюда следует, что
dU = TdS – pdV (4 – 1)
Введя новые функции состояния
U ≡ U + pV , G ≡ H – TS , F ≡ U — TS
можно получить четыре эквивалентные формы записи:
 dU = TdS – pdV
dU = TdS – pdV
dF = — pdV – SdT (4 — 2)
Все выражения (4 – 2) справедливы лишь для закрытых систем, в которых не совершаются другие виды работ за исключением механической работы. В этом случае, как уже отмечалось
U = U ( S , V ) H = H ( S , P ) G = G ( P , T ) F = ( V , T ) являются термодинамическими потенциалами и определяют направления самопроизвольного протекания процесса. Но, совершенно очевидно, что эти процессы связаны либо с выделением энергии в форме теплоты, либо с совершением механической работы. Никакие другие процессы здесь не рассматриваются.
Если в системе протекает химическая реакция, она становится аналогом открытой системы, поэтому возникает необходимость учитывать обмен энергией и массой с окружающей средой. Поэтому термодинамические потенциалы будут функцией всех веществ участвующих в реакции и систему уравнений (4 – 2) следует записать:





Все производные по количеству вещества равны между собой и характеризуют приращение соответствующего термодинамического потенциала при измерении данного вещества при фиксированных естественных переменных и неизменных количествах остальных веществ: (4 — 4)
µ i = (  )S,V, nj≠i = (
)S,V, nj≠i = (  ) S,P,nj≠i = (
) S,P,nj≠i = (  ) V,T,nj≠i = (
) V,T,nj≠i = (  )P,T,nj≠i
)P,T,nj≠i
Эта новая функция, введенная Гиббсом, получила название химического потенциала. Химический потенциал фактически является движущей силой при массопереносе. Система самопроизвольно переходит в состояние с более низшим потенциалом. Более нагретое тело, с большей температурой, нагревает менее нагретое тело, с меньшей температурой, т.е. температура – потенциал при теплопередачи, т.е. температура выступает здесь движущей силой. Работа расширения совершается аналогично за счет уменьшения давления: давление потенциал в случае механической работы. Все потенциалы, как известно интенсивные переменные, т.е. не зависящие от массы. Любой самопроизвольный процесс – это процесс приводящий к выравниванию потенциалы в результате чего достигается равновесие.
Очевидно, что если в системе совершаются другие виды работ, связанные с переносом массы, то учет ∑μ idni не достаточен.
Уравнения (4 -3) справедливы лишь в том случае, если химическая реакция протекает в гомогенной системе и системе не совершает других видов работ, кроме механической. Очевидно в этом случае:
 dU =
dU =  μidni (S,V = const)
μidni (S,V = const)
dH = μidni (S, p = const)
dG = μidni (T, p = const)
dF = μidni (T,V = const)
Для гетерогенной системы эти уравнения должны быть записаны для каждой фазы в отдельности. При этом, равновесие в гетерогенной системе наступает лишь тогда, когда химические потенциалы всех компонентов во всех фазах выравниваются.
Среди рассмотренных термодинамических потенциалов в конкретных термодинамических расчетах наибольшее значение имеют энергия Гельмгольца F и энергия Гиббса G . Их естественные переменные наиболее удобны, поскольку легко реализуемы. Изменение этих функций в каком – либо процессе при постоянстве естественных переменных равно максимальной работе, которая система может совершить в этом процессе при проведении процесса обратимо. Изменение этих термодинамических потенциалов можно найти из термодинамических констант, поэтому они позволяют определить возможность самопроизвольного процесса и глубину его протекания:
∆ GT = ∆ HT — T ∆ ST (4 – 6)
∆ FT = ∆ HT — T ∆ ST (4 – 7)
Если условия протекания процесса не сильно отличаются от стандартных, то в расчете можно использовать термические константы, отнесенные к стандартным условиям. В этом случае расчет ∆ rG ° T может быть осуществлен:
где ∆ rH º T и ∆ rS º T рассчитываются на основе стандартных энтальпий образования и абсолютных энтропий с пересчетом к температуре T .
Изменения энергии Гиббса может быть рассчитано аналогично тепловому эффекту реакции с использованием энергий Гиббса образования:
∆ rG ° T = ∑ υi ∆ fG ° T — ∑ υj ∆ fG ° T (4 – 8)
Рассмотрим равновесия типа:

 Жидкость Пар
Жидкость Пар
Твердое тело Пар

 Твердое тело Жидкость
Твердое тело Жидкость
Равновесие, как известно, характеризуется равенством энергий Гиббса для индивидуального вещества.
Исходя из (4 – 2) для жидкой и паровой фаз мы можем записать:
 dG ж = V ж d р – S ж dT
dG ж = V ж d р – S ж dT
dG пар = V пар d р — S пар dT (4 – 9)
Если энергии Гиббса отнесены к 1 молю, то условие равновесия может быть записано:
Отсюда следует, что
Из системы (4 – 9) получаем:
V ж dp – S ж dT = V п dp – S п dt
Отсюда следует, что
 (4 – 10)
(4 – 10)
Уравнение (4 – 10) – это уравнение Клаузиуса – Клайперона, которое показывает, как изменяется давление с ростом температуры при изменении агрегатного состояния вещества.
Уравнение (4 – 10) применимо к любому фазовому переходу I рода. Оно может быть преобразовано с учетом того, что
∆ G ф.п. = ∆ H ф.п . — Т∆ S ф.п. = 0
Для процессов испарений и сублимации V тв/ж ›› V п. Считая, что пар подчиняется закону идеальных газов: V п = RT / P . Уравнение (4 – 10) можно преобразовать:
 (4 — 10′)
(4 — 10′)
Уравнения (4 – 10) и (4 – 10 ′ ) позволяют определить энтальпию фазового перехода по температурной зависимости давления паров. В случае плавления требуются дополнительные сведения о больших объемах жидкой и твердой фаз.
Рассмотрим несколько примеров таких расчетов.
Приняв энтальпию плавления льда при 273,15 К равный 333,5 Дж∙2ˉ¹, а удельные объемы жидкой воды и льда равными 1,0002 см³∙2ˉ¹ и 1,0908 см³∙2ˉ¹ соответственно, рассчитать на сколько нужно повысить давление, чтобы температура плавления льда уменьшилась на 1 К.
Из уравнения (4 – 10) следует 


Знак «-» указывает, что температура плавления льда уменьшается с ростом давления.
При давлении 1 атм. Бензол кипит при 80,1º С. Какое давление паров бензола при стандартной термодинамической температуре?
На основании уравнения (4 — 10′) получаем
 │
│ 
В условии задачи отсутствует ∆Н исп . Её можно найти через энтальпии образования, предположив, что энтальпия испарения слабо зависит от температуры.
 = 82,98 кДж∙мольˉ¹
= 82,98 кДж∙мольˉ¹
 = 49,07 кДж∙мольˉ¹
= 49,07 кДж∙мольˉ¹
Следовательно, ∆ бар Нº(298, С6Н6) = 33,91 кДж∙мольˉ¹


Возможна и другая оценка энтальпии испарения. Для неполярных жидкостей выполняется правило Трутона, согласно которому мольная энтальпия испарения в точке НТК (нормальной температуры кипения) примерно равна 88 Дж∙мольˉ¹ Кˉ¹.
Оценивая для бензола ∆Н исп., получаем ∆Н исп. = 88∙353,3 = 31100 Дж∙мольˉ¹. В этом случае получаем р=0,141 атм.
Какое давление паров бензола при стандартной термодинамической температуре, если стандартные энергии Гиббса образования жидкого и газообразного бензола равны +124,43 кДж∙мольˉ¹ и +129,75 кДж∙мольˉ¹ соответственно.
Если Т = const = 298,15 то

Принимая ∆ V = V пар = получаем, после интегрирования



При каком давлении алмаз и графит находятся в равновесии друг с другом при температуре 298,15 К, если плотность графита и алмаза 2,25 и 3,51 г∙смˉ³ соответственно, а  = 2,900 кДж∙мольˉ¹
= 2,900 кДж∙мольˉ¹
 Сграфит Салмаз
Сграфит Салмаз

∆ fG =2,900 кДж∙мольˉ¹
Поскольку  , то
, то 
По условию равновесие соответствует ∆G2 = 0, следовательно
P 2 – P 1 = ∆G2 — 
∆ G 1= — ∆ fG º. Если принять, что стандартные условия отнесены к Р1 = 1 атм. = 101325,1 Па,
то  =
= 
При 70ºС давление насыщенных паров CCl 4 621,15 мм рт. ст, а при 80ºС 843,29 мм рт. ст. Чему равна энтальпия испарения СС l .
Из уравнения Клаузиуса – Клайперона
 , получаем
, получаем
 │=
│=  │=
│= 
ОБЪЕДИНЕННОЕ УРАВНЕНИЕ ПЕРВОГО И ВТОРОГО ЗАКОНОВ ТЕРМОДИНАМИКИ
При термодинамических исследованиях, ориентированных на решение различных практических задач, определение направления и пределов течения процессов, может быть применен второй закон термодинамики и использована функция энтропия. Однако положения второго закона термодинамики и поведение функции 5 обычно рассматриваются в применении к изолированным системам. На практике приходится иметь дело с системами, взаимодействующими с окружающей средой и в этом случае для характеристики системы и выявления характера процессов, протекающих в ней, требуются другие функции. Такие функции и соответствующий метод исследования получены благодаря работам Гиббса, Максвелла, Масье, Гельмгольца и др. Эти функции называются характеристическими функциями (у Гиббса они называются фундаментальными).
Характеристической функцией называется функция состояния независимых параметров, характеризующаяся тем, что посредством этой функции и ее производных по этим параметрам могут быть выражены все термодинамические свойства системы.
К характеристическим функциям относятся: внутренняя энергия U, энтальпия Н, энергия Гельмгольца (изохорно-изотермический потенциал) F, энергия Гиббса (изобарно-изотермический потенциал) G и энтропия S.
Дифференциалы этих функций являются полными, функции аддитивны. Каждой функции соответствует вполне определенная пара постоянных параметров (если за независимые параметры принимать другие параметры, то данная функция от этих переменных не будет характеристической).
Метод исследования с помощью характеристических функций базируется на применении объединенного уравнения первого и второго законов термодинамики для обратимых процессов.
Из уравнений первого и второго законов термодинамики δQ = = dU + δА и dS = δQ/T можно получить следующие:
 (86)
(86)
Если в системе совершается работа только против сил внешнего давления, то уравнения (86) примут вид
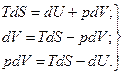 (88)
(88)
Уравнения (88) являются объединенными уравнениями первого и второго законов термодинамики. Каждое из этих уравнений связывает между собой пять переменных величин (Т, S, U, р и V), характеризующих состояние системы.
Если две из указанных величин принять за независимые переменные (например, Т и р или T и V), то в уравнении останется три неизвестных величины. Для решения вопроса о состоянии системы необходимо к написанному уравнению добавить еще два. Одно из этих уравнений может быть уравнением состояния f(р, V, Т) для идеальных газов —это pVμ = RT, а второе —какое-либо калорическое уравнение.
Объединенное уравнение для I и II законов термодинамики.
Правило Гесса.
Протекание химических реакций сопровождается перераспределением электронов в молекулах (атомах, ионах и т. д.), что вызывает изменение внутренней энергии и энтальпии системы. Изменение энергии может сопровождаться поглощением или выделением теплоты, а также совершением работы.
Тепловой эффект реакции отождествляется с изменением внутренней энергии для систем c V = const : Qv = ΔU и с изменением энтальпии для систем с р = const: Qp = ΔH.
Правило Гесса устанавливает, что тепловой эффект химической реакции, протекающей при постоянном объеме или постоянном давлении, не зависит от промежуточных реакций, а определяется лишь начальным и конечным состоянием реагирующих веществ.
Во всех остальных случаях правило Гесса недействительно.
Зависимость теплового эффекта от температуры определяется по уравнению Кирхгофа.
Изменение энтальпии реакции (или произвольного процесса) при постоянном давлении и повышении температуры на один градус равно сумме теплоемкостей продуктов реакции (процесса), умноженных на стехиометрические коэффициенты, за вычетом теплоемкостей исходных веществ, также умноженных на стехиометрические коэффициенты.
 , после интегрирования: ΔHТ = ΔHО +
, после интегрирования: ΔHТ = ΔHО +  (4)
(4)
Где a, b, c, d – стехиометрические коэффициенты реакции
Пример. В качестве противопригарной добавки в формовочную смесь вводится марганцевая руда. Для определения количества руды в смеси необходимо знать изменение энтальпии в результате взаимодействия закиси марганца с кварцевым песком при заливке формы металлом при 1500° К:

Теплоемкость компонентов реакции находим из справочника

Изменение энтальпии реакции определим по формулам (2) и (4). При расчете сделаем допущение, что каждый из компонентов реакции не испытывает превращений в твердой фазе до температуры 15006 К (на самом деле .кремнезем и закись марганца испытывают ряд фазовых превращений до этих температур, но теплоты этих превращений малы и в первом приближении их можно не учитывать). В этом случае
После подстановки числовых значений теплоемкости и стандартных энтальпий получаем

ΔН о т= 1240,3—37,143 = 1203,157 кдж/моль.
ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ
Этот закон устанавливает определенные закономерностей превращения теплоты в работу и обратно. Являясь двумя формами передачи энергии, теплота и работа не являются равноценными: если работа может непосредственно пойти на увеличение любого вида энергии, то теплота непосредственно, без превращения ее в работу, приводит лишь к увеличению внутренней энергии системы.
Второй закон позволяет разделить все процессы на два вида:
обратимые и необратимые. Мерой необратимости процесса является энтропия – S – параметр состояния тела, полный дифференциал которой меньше, либо равен отношению бесконечно малого приращения затраченной теплоты к температуре процесса: dS = 
где  — приращение энтропии;
— приращение энтропии;  — минимальная теплота, подведенная к системе; T — абсолютная температура процесса;
— минимальная теплота, подведенная к системе; T — абсолютная температура процесса;
Для обратимого кругового процесса изменение энтропии = нулю (те. сама энтропия является постоянной величиной)
Для необратимого кругового процесса изменение этропии всегда положительно
Соответственно, dS ≥ 0 – II закон термодинамики.
При стремлении системы к равновесию
Энтропия является аддитивной величиной — энтропия сложной системы
равна сумме энтропии ее отдельных частей.
Энтропия отдельных частей системы в произвольном процессе
может как возрастать, так и уменьшаться (при возрастании энтропии системы в целом). При любом процессе, ускоряющем движение частиц, энтропия возрастает (плавление, испарение, диффузия и т. д.).
Изменение энтропии при произвольном превращении равно
разности энтропии конечного и начального состояний, т. е.

Объединенное уравнение для I и II законов термодинамики.
Решение системы уравнений (1 з-н ) и (2. з-на) позволяет написать объединенное уравнение для первого и второго законов термодинамики для любого компонента системы:
Из последнего уравнения следует, что внутренняя энергия является функцией двух независимых переменных — энтропии и объема, т. е.
Таким образом, U – внутренняя энергия – является характеристической функцией, так как с помощью ее производных можно выразить в явной форме термодинамические параметры системы. Например,
P =- (  )S и T = (
)S и T = (  )V;
)V;
Таким образом, давление в системе можно выразить как взятую с обратным знаком скорость изменения внутренней энергии по объему при постоянной энтропии системы. Температура в системе равна скорости изменения внутренней энергии по энтропии при постоянном объеме.
Обладая запасом энергии U, система не может эту энергию полностью перевести в полезную работу.
Внутреннюю энергию системы можно представить в виде суммы:
где F – cвободная энергия, которая может быть использована для производства работы (ее также называют — св. энергия Гельмгольца, свободная энергия при постоянном объеме, изохорно-изотермический потенциал.);
TS – связанная энергия, которая безвозмездно теряется в процессе.
F – свободная энергия , тоже характеристическая функция, так как с ее помощью можно определить в явной форме ряд термодинамических параметров системы.:
Таким образом, свободная энергия является функцией двух независимых переменных Т и V. Полный дифференциал функции F можно выразить следующим образом:


Эти соотношения позволяют, например, рассчитать давление в системе, если известна величина свободной энергии и ее зависимость от объема.
Условием равновесия для изохорно-изотермического процесса является условие минимума функции F при равновесии
Работа при изохорно-изотермическом процессе равна разности свободных энергий:
ΔF позволяет судить о направлении процессов в незамкнутых системах, т.к. характеризует максимальную полезную работу.
Для изобарно-изотермического процесса введена функция – свободная энтальпия или термодинамический потенциал Гиббса (св. энергия Гиббса):
G = U + pV — TS = Н — TS.
Полное значение энтальпии можно представить в виде суммы
двух частей:
Н = G + TS = (Н — TS) + TS.
Первая часть характеризует ту энергию, которая может быть использована в процессе (свободная энтальпия), а вторая — энергию, которую невозможно использовать в изобарно-изотермическом процессе (связанная энтальпия).
Так же как и свободная энергия, свободная энтальпия является термодинамическим потенциалом. В дифференциальной форме уравнение свободной энтальпии имеет вид:
dG = — S dT + V dp
Таким образом, свободная энтальпия является функцией двух независимых переменных — температуры и давления.
При постоянной температуре и давлении изменение свободной энтальпии равно нулю, т.е. Т=const, P=const ΔG=0
Полный дифференциал свободной энтальпии можно представить в виде:
dG=  , имея в виду предыдущее выражение, можно записать:
, имея в виду предыдущее выражение, можно записать:
V= 
S= 
Используя эти выражения, зная экспериментальную зависимость свободной энтальпии G от температуры, можно найти значение эН для системы и ΔН для процесса:
Н=G – T
ΔН=ΔG –  Эти два выражения называются уравнениями Гиббса.
Эти два выражения называются уравнениями Гиббса.
При самопроизвольном течении изобарно-изотермического процесса свободная энтальпия системы снижается. Для неравновесного процесса dG ≤0 — условие минимума функции G.
Для равновесного процесса dG = 0.
Следовательно, при стремлении процесса к равновесию свободная энтальпия стремится к Gmin, и при равновесии она достигает минимального значения.
Для изобарно-изотермического процесса свободная энтальпия связана с работой:
Ар,Т = АV,Т – pΔV≤G1 – G2
Это означает, что термодинамическая функция G может служить критерием направленности протекания изобарно-изотермических процессов.
Большинство реальных процессов протекает между различными частями системы с переменным числом частиц. Изменение числа частиц в частях системы может быть вызвано различными причинами: механическими, химическими, электромагнитными и т. д.
Общее число частиц в системе при этом может изменяться (большинство химических реакций) или оставаться неизменным (плавление, испарение).
Перемещающиеся частицы несут определенную энергию, которую необходимо учитывать при расчете равновесного состояния системы.
Рассмотрим произвольную систему с переменным числом частиц, но при условии, что ∑ni= 1 молю. В этом случае вместо числа молей ni нужно писать мольную долю Ni. Для каждой частицы справедливы законы термодинамики. Поэтому в термодинамические уравнения необходимо ввести член, учитывающий перемещения частиц вещества:
dF= – SdT – pdV+∑μidNi (изохорно-изотермический)
dG= – SdT + Vdp+∑μidNi (изобарно-изотермический)
где μi – коэффициент пропорциональности:

Коэффициент μi называется химическим потенциалом компонента и является мерой изменения характеристических функций при перемещении частиц из одной части системы в другую. Наибольшее распространение получило выражение химического потенциала через свободную энтальпию, так как все характеристические функции, кроме AG, зависят от числа частиц в системе.
Для одной ‘частицы

G = f4 (p, Т).
Выражение химического потенциала через ΔG:

Численно μiравно изменению свободной энтальпии на одну частицу или на моль вещества.
В дифференциальной форме уравнение для химического потенциала имеет вид:
dμ = — s dT + v dp,
где s —энтропия одной частицы (моля);
v — парциальный объем компонента.
 |
Химический потенциал позволяет определить влияние внешних факторов на процессы, протекающие в системе. Все самопроизвольные процессы идут в направлении уменьшения химического потенциала. При этом должно выполняться соотношение:
Последнее выражение называется уравнением Гиббса — Дюгема. Оно особенно удобно для исследования фазовых превращений.
Химический потенциал компонента вещества в системе можно
вычислить следующим образом:

где μi° (N, Т) — химический потенциал чистого вещества при данной температуре, давлении и агрегатном состоянии в системе.
Или,

где μi ° (Т, ро) — химический потенциал i-ro компонента в фазе при постоянной температуре и давлении р0 = 1;
μi ° (Т, с0) — химический потенциал i-ro компонента в фазе при постоянной температуре и концентрации с0=1.
Эти уравнения химического потенциала справедливы лишь для идеальных смесей (твердых, жидких или газообразных). В идеальных растворах (и совершенных – концентрированных и разбавленных) частицы растворенного вещества и растворителя не взаимодействуют друг с другом.
ТЕПЛОВАЯ ТЕОРЕМА НЕРНСТА
Второй закон термодинамики позволяет определить вероятность произвольного процесса, проходящего в исследуемой системе.
Однако для вычисления полноты этого процесса необходимо знать абсолютные значения термодинамических величин, рассчитать которые на основании второго закона термодинамики невозможно.
Числовые значения энтропии, энтальпий, свободных энергий и констант равновесия рассчитывают с точностью до постоянной интегрирования. При этом необходимо, кроме температурных зависимостей термодинамических функций, знать их числовые значения при строго определенных условиях.
Исследования, проведенные В. Нернстом, позволили определить постоянную интегрирования. Опираясь на богатый экспериментальный материал, накопленный при изучении поведения веществ при низких температурах, В. Нернст установил, что разность свободной и внутренней энергии при понижении температуры стремится к нулю быстрее, чем по линейному закону, т. е. в уравнении ΔU — ΔF = TΔS при Т — 0 член ΔS также стремится к нулю. Это значит, что lim ΔS = 0.
Таким образом, при всех изменениях, происходящих в чистых конденсированных веществах при абсолютном нуле, изменение энтропии равно нулю. Последнее утверждение является тепловой теоремой Нернста.
М. Планк предположил, что при абсолютном нуле не только изменение энтропии равно нулю, но и сама энтропия равна нулю (постулат Планка), т. е. ΔS = 0 при Т → 0 и S = 0 при Т — 0.
Равенство энтропии нулю при Т → 0 связано с квантовой природой реальных систем. При абсолютном нуле система должна находиться в единственно возможном состоянии с минимальной энергией. Для ряда веществ (сплавы, аморфные тела, химические соединения), как показывают измерения, энтропия стремится не к нулю, а к некоторой положительной величине. Однако это указывает не на ограниченность постулата Планка, а на то, что указанные вещества не находятся в равновесных состояниях. Cсправедливость теоремы Нернста и постулата Планка принимается для всех систем.
Теорема Нернста и постулат Планка позволяют определять абсолютные значения термодинамических величин при произвольных температурах:


УСЛОВИЯ РАВНОВЕСИЯ ТЕРМОДИНАМИЧЕСКОЙ СИСТЕМЫ
Общие условия равновесия произвольных систем в следующих процессах:




При расчете равновесий чаще всего выбирают изохорно-изотермические или изобарно-изотермические процессы.
Равновесие в однородной среде.
Условия равновесия в однородной среде (которая по определению не имеет поверхности раздела) следуют из уравнений . Это равенство температур, давлений, концентраций, химических потенциалов, всех видов энергий во всех точках системы (при отсутствии внешнего поля).
Равновесие в гетерогенной системе.
Химический потенциал произвольного компонента в системе при равновесии должен быть одинаков во всех фазах независимо от числа фаз и компонентов системы. Химический потенциал какого-либо вещества в различных агрегатных состояниях также должен быть одинаков во всех фазах:
Это позволяет определить химический потенциал произвольного вещества по парциальному давлению его паров:
Уравнение Клаузиуса — Клапейрона.
Процессы изменений фазового состояния вещества, протекающие без химического взаимодействия, называются фазовыми превращениями. Основными характеристиками фазовых превращений являются температура и давление, взаимосвязанные между собой.
Зависимость температуры превращения от давления можно определить из условия равновесия химических потенциалов при равновесии двух фаз:
μ I (p, T) = μ II (p, T)
Дифференцируя это уравнение, получим
dμ I (p, T) = dμ II (p, T)
или, учитывая, что dμ является полным дифференциалом: 
 это выражение называется уравнением Клаузиуса—Клапейрона. Его можно записывают через теплоту фазового превращения:
это выражение называется уравнением Клаузиуса—Клапейрона. Его можно записывают через теплоту фазового превращения:

где Qnp = Т (s» — s’) = TΔS — теплота фазового превращения;
v» — v’ — изменение объема при фазовом превращении.
Для плавления Q = λпл, λпл = Тпл 
Для испарения Q = рис, pисп = Тисп 
Из уравнения Клаузиса—Клапейрона следует, что любое превращение в системе, связанное с изменением объема или плотности, имеет температуру превращения, зависящую от давления.
При плавлении металлов и сплавов Тпл зависит от давления в незначительной степени. Ниже показана зависимость температуры плавления свинца от внешнего давления:
| Р, Мн/м 2 | 0,1 | 25,0 | 50,0 | 100,0 | 200,0 |
| Тпл, °К | 623,51 | 625,62 | 627,36 | 631,44 | 639,54 |
В общем виде уравнение Клаузиуса—Клапейрона не решается, из-за сложной зависимости от температуры.
Однако его можно решить, сделав ряд допущений.
Рассмотрим процесс испарения или разложения конденсированного вещества (твердого или жидкого) с выделением из него газов.
Допустим: 1) объемом конденсированной фазы по сравнению
с газообразной можно пренебречь, т.е. ΔV = Vгаз—Vконд = Vгаз
2) газ подчиняется уравнению состояния идеальных газов, т. е. для 1 моля
V = 
Учитывая допущения, общее решение уравнения Клаузиуса — Клапейрона:
ln 
Рассмотрим 3 случая:
1. Энтальпия превращения не зависит от температуры. В этом случае

Или, учитывая значения R=8,314 и модуль перехода ln→lg=2,303

2. Энтальпия превращения не зависит от температуры в интервале температур T1 — T2.
В этом случае исследуемый участок температур разбивают на ряд интервалов с ΔHi = const и общее уравнение интегрируют по частям. В результате получают:
lg 
3. Зависимость энтальпии превращения от температуры в общем случае выражается уравнением:
 ΔH = ΔH o + αT + βT 2 +…
ΔH = ΔH o + αT + βT 2 +…
В этом случае решение следующее:
lgp = 
Плавка чистого магния сопровождается обильным паровыделением легко воспламеняющегося магния (горит в присутствии кислорода воздуха) Чтобы уменьшить испарение магния, его поверхность покрывают слоем шлака. Эффективность такой защиты сохраняется только до температуры кипения магния. Экспериментально установлена зависимость давления паров магния от температуры:
ln pMg = 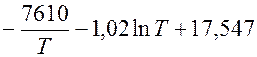
Определить температуру кипения Mg при р = 100 кн/м 2 (760мм рт.ст.)
Температуру кипения Mg при р=100 кн/м 2 определим по экспериментальной зависимости:
ln 100 =
1,02 Т lnТ – 12,941Т + 7610 = 0
Ткип = 1376ºК (1103ºС)
Правило фаз.
Рассмотрим систему из K независимых компонентов, размещенных в Ф фазах. Химический потенциал каждого компонента μк ф , где нижний индекс обозначает компонент, верхний ‒ фазу.
Состояние гетерогенной системы определяется температурой, давлением и концентрацией.
Для определения концентрации каждого К-го компонента в какой-либо фазе, необходимо знать содержание (К -1) компонентов в той же фазе. Таким образом, для описания состояния системы необходимо решить Ф×(К-1) уравнений.
Однако общее число уравнений может быть сокращено, если учесть, что химический потенциал i-го компонента во всех фазах при равновесии должен быть одинаков:

Таким образом, число независимых уравнений Ф×(К-1) может быть сокращено на К×(Ф-1) уравнений.
Т.О. Число независимых уравнений для определения концентрации каждого компонента равно:
Число независимых уравнений состояния термодинамической системы равно числу термодинамических степеней свободы си указывает число независимых параметров, изменение которых (в определенных пределах) не изменяет числа фаз и компонентов системы.
Найденное нами число степеней свободы не учитывает влияние внешних параметров, включая р и Т. По второму постулату термодинамики внутренние параметры системы являются функцией внешних параметров и температуры. Следовательно, к числу степеней свободы необходимо добавить число неучтенных внешних параметров:
Это выражение называется уравнением Гиббса—Коновалова.
В металлургии, литейном производстве, материаловедении обычно учитывают р и Т или только Т.
РАВНОВЕСИЕ ХИМИЧЕСКИХ РЕАКЦИЙ
Рассмотрим равновесие химической реакции догорания окиси углерода:
2СО + О2 ↔2СО2; ΔН° = —566 200 дж/моль.
Равновесие всей системы при Т = const, p — const определяется из условия минимума свободной энтальпии, G→min:
В данной системе все вещества взаимно растворены, поэтому при расчетах необходимо учитывать энтропию смешения:
Ni — мольные доли компонентов в системе.
При постоянной температуре ΔH° и ΔS o — величины постоянные. Следовательно, и сумма энтропии смешений при постоянной температуре должна быть величиной постоянной, т. е.
Ln  = const,
= const,
=KN = const.
Величина KN называется константой равновесия, выраженной через мольные доли компонентов реакции. При постоянных температуре и давлении константа равновесия не зависит от количественного соотношения участвующих в реакции веществ. Если изменить концентрацию одного из компонентов реакции, то изменятся концентрации всех других веществ, но таким образом, что константа равновесия останется без изменения.
Для произвольной реакции
аА + bВ +—- ↔ сС + dD + —-
константа равновесия равна
KN = 
где vi; — стехиометрические коэффициенты реакции;
П — знак произведения.
Знание величины константы равновесия позволяет определить
полноту протекания химических реакций.
При К > 1 — реакция смещается вправо;
K > 1 — система состоит в основном из продуктов реакции;
К 0, то это значит, что реакция идет в обратном направлении, (справа налево), т.е.в сторону образования исходных веществ.
2) рассчитываются энергии и энтропии взаимодействия для заданных условий;
3) рассчитываются константы равновесия Кр.
Сначала проводят предварительный расчет, определяя ΔG o T(ΔF o T) или пренебрегая изменением теплоемкости во время реакции (ΔСр = 0), или считают это изменение постоянным, не зависящим от температуры, ΔСр =const.
Если получают ΔG o T(ΔF o T) o T(ΔF o T)>>0, то реакция практически не идет.
В этих случаях надобность в точном расчете отпадает.
Расчет проводят при отсутствии фазовых превращений в изохорно-изотермической системе по выражению:
 (1)
(1)


Если ΔCp = const, то

Итак, приблизительный расчет выполнили, решили, что точный расчет необходим. Проводим его по уравнению (1), преобразовав в более удобную форму:

Значения двойных интегралов находят в справочниках для различных значений температур.
В справочниках приведены значения коэффициентов разложения этих двойных интегралов в ряд по методу Темкина-Шварцмана:
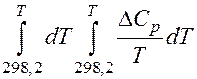 = ΔaMo+ ΔbM1+ ΔcM2+ ΔdM3+ Δc΄M-2+…
= ΔaMo+ ΔbM1+ ΔcM2+ ΔdM3+ Δc΄M-2+…
где Δa(Δb, Δc, Δc΄, Δd) = 
Vj — коэффициенты уравнений теплоемкости реагирующих веществ;

Значения коэффициентов М0, M1, М2, М3, М-2 приведены в справочниках для различных значений температур.
С учетом указанного разложения уравнение для вычисления
свободной энтальпии записывается в виде:
 T( ΔaMo+ ΔbM1+ ΔcM2+ ΔdM3+ Δc΄M-2)
T( ΔaMo+ ΔbM1+ ΔcM2+ ΔdM3+ Δc΄M-2)
Далее вычисляют K по уравнению:
lnK =  и по ее величине и знаку судят о направлении и полноте реакции.
и по ее величине и знаку судят о направлении и полноте реакции.
Следует знать, что связь константы равновесия со свободной энтальпией — одна из наиболее важных зависимостей в термодинамике. Небольшая ошибка в определении ΔG повлечет за собой принципиально неверное значение K.
Наиболее часто встречающееся значение свободной энтальпии равно примерно 200 кдж/моль. Таким образом, ошибка 4% в величине ΔG может привести к ошибке 100% при определении К.
Поэтому ясно, что необходима исключительная точность при вычислении свободной энтальпии, если ее используют для расчетов константы равновесия.